Expression
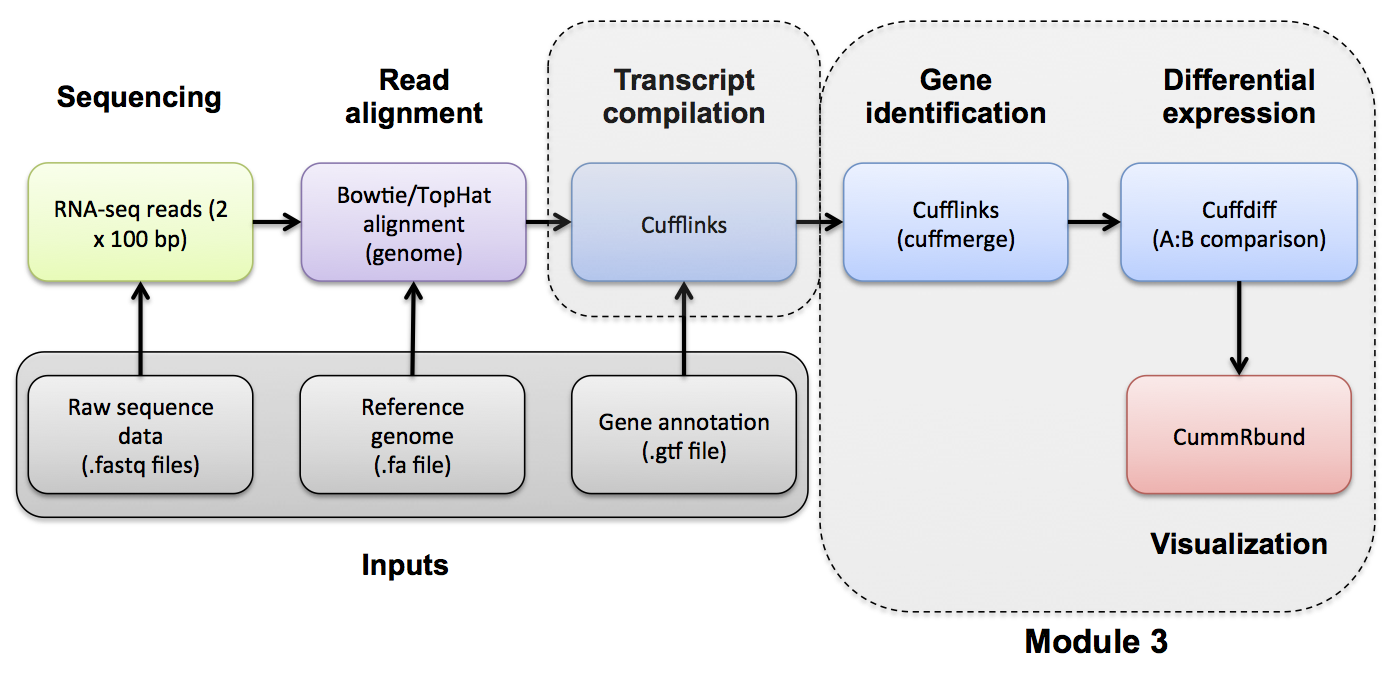
#3-i. Expression Use Cufflinks to generate expression estimates from the SAM/BAM files generated by TopHat in the previous module
###Note on de novo transcript discovery and differential expression using Cufflinks and Cuffdiff In this module, we will run Cufflinks in 'reference only' mode. For simplicity and to reduce run time, it is sometime useful to perform expression analysis with only known transcript models. However, Cufflinks can predict the transcripts present in each library instead (by dropping the '-G' option in cufflinks commands as described later in Module 5). Cufflinks will then assign arbitrary transcript IDs to each transcript assembled from the data and estimate expression for those transcripts. One complication with this method is that in each library a different set of transcripts is likely to be predicted for each library. There may be a lot of similarities but the number of transcripts and their exact structure will differ in the output files for each library. Before you can compare across libraries you therefore need to determine which transcripts correspond to each other across the libraries.
- Cufflinks provides 'cuffmerge' to combine predicted transcript GTF files from across different libraries
- Once you have a merged GTF file you can run Cuffdiff with this instead of the known transcripts GTF file we used above
- Cufflinks also provides 'cuffcompare' to compare predicted transcripts to known transcripts
- Refer to the Cufflinks manual for a more detailed explanation:
- http://cole-trapnell-lab.github.io/cufflinks/manual/
Cufflinks basic usage:
#cufflinks [options] <hits.sam>
Extra options specified below:
'-p 8' tells Cufflinks to use eight CPUs '-G/--GTF ' Forces cufflinks to calculate expression values for just known transcripts (we call this "reference only" mode) '--library-type fr-firststrand' Tells Cufflinks we are analyzing a strand-specific library protocol but is not required since TopHat provides the XS BAM tag. '-o' tells Cufflinks to write output to a particular directory (one per sample)
cd $RNA_HOME/
mkdir -p expression/tophat_cufflinks/ref_only/
cd expression/tophat_cufflinks/ref_only/
cufflinks -p 8 -o HBR_Rep1_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/HBR_Rep1_ERCC-Mix2/accepted_hits.bam
cufflinks -p 8 -o HBR_Rep2_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/HBR_Rep2_ERCC-Mix2/accepted_hits.bam
cufflinks -p 8 -o HBR_Rep3_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/HBR_Rep3_ERCC-Mix2/accepted_hits.bam
cufflinks -p 8 -o UHR_Rep1_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/UHR_Rep1_ERCC-Mix1/accepted_hits.bam
cufflinks -p 8 -o UHR_Rep2_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/UHR_Rep2_ERCC-Mix1/accepted_hits.bam
cufflinks -p 8 -o UHR_Rep3_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/tophat/UHR_Rep3_ERCC-Mix1/accepted_hits.bam
###OPTIONAL ALTERNATIVE - Cufflinks on STAR Run cufflinks on STAR alignments instead of TopHat alignments. Note, the library type is now required since STAR does not produce the appropriate XS tags for strand-specific RNA-seq protocols:
cd $RNA_HOME/
mkdir -p expression/star_cufflinks/ref_only/
cd expression/star_cufflinks/ref_only/
cufflinks -p 8 -o HBR_Rep1_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/HBR_Rep1/Aligned.out.sorted.bam
cufflinks -p 8 -o HBR_Rep2_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/HBR_Rep2/Aligned.out.sorted.bam
cufflinks -p 8 -o HBR_Rep3_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/HBR_Rep3/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep1_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/UHR_Rep1/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep2_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/UHR_Rep2/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep3_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/star/UHR_Rep3/Aligned.out.sorted.bam
###OPTIONAL ALTERNATIVE - Cufflinks on HISAT2 Run cufflinks on HISAT2 alignments instead of TopHat alignments.
cd $RNA_HOME/
mkdir -p expression/hisat2_cufflinks/ref_only/
cd expression/hisat2_cufflinks/ref_only/
cufflinks -p 8 -o HBR_Rep1_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/HBR_Rep1/Aligned.out.sorted.bam
cufflinks -p 8 -o HBR_Rep2_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/HBR_Rep2/Aligned.out.sorted.bam
cufflinks -p 8 -o HBR_Rep3_ERCC-Mix2 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/HBR_Rep3/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep1_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/UHR_Rep1/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep2_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/UHR_Rep2/Aligned.out.sorted.bam
cufflinks -p 8 -o UHR_Rep3_ERCC-Mix1 --library-type fr-firststrand --GTF $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/alignments/hisat2/UHR_Rep3/Aligned.out.sorted.bam
What does the raw output from Cufflinks look like?
cd $RNA_HOME/expression/tophat_cufflinks/ref_only/UHR_Rep1_ERCC-Mix1/
ls -l
head isoforms.fpkm_tracking
cut -f 1,4,7-13 isoforms.fpkm_tracking | less
Press 'q' to exit the 'less' display
###OPTIONAL ALTERNATIVE - HTSEQ-COUNT Run htseq-count on alignments instead to produce raw counts instead of FPKM values for differential expression analysis
Refer to the HTSeq documentation for a more detailed explanation:
htseq-count basic usage:
htseq-count [options] <sam_file> <gff_file>
Extra options specified below:
- '--format' specify the input file format one of BAM or SAM. Since we have BAM format files, select 'bam' for this option.
- '--order' provide the expected sort order of the input file. Previously we generated position sorted BAM files so use 'pos'.
- '--mode' determines how to deal with reads that overlap more than one feature. We believe the 'intersection-strict' mode is best.
- '--stranded' specifies whether data is stranded or not. The TruSeq strand-specific RNA libraries suggest the 'reverse' option for this parameter.
- '--minaqual' will skip all reads with alignment quality lower than the given minimum value
- '--type' specifies the feature type (3rd column in GFF file) to be used. (default, suitable for RNA-Seq and Ensembl GTF files: exon)
- '--idattr' The feature ID used to identity the counts in the output table. The default, suitable for RNA-SEq and Ensembl GTF files, is gene_id.
We could use either the tophat, HISAT2, or STAR alignments. We have chosen to use tophat alignments here. Run htseq-count and calculate gene-level counts:
cd $RNA_HOME/
mkdir -p expression/tophat_counts
cd expression/tophat_counts
mkdir UHR_Rep1_ERCC-Mix1 UHR_Rep2_ERCC-Mix1 UHR_Rep3_ERCC-Mix1 HBR_Rep1_ERCC-Mix2 HBR_Rep2_ERCC-Mix2 HBR_Rep3_ERCC-Mix2
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/UHR_Rep1_ERCC-Mix1/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > UHR_Rep1_ERCC-Mix1/gene_read_counts_table.tsv
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/UHR_Rep2_ERCC-Mix1/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > UHR_Rep2_ERCC-Mix1/gene_read_counts_table.tsv
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/UHR_Rep3_ERCC-Mix1/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > UHR_Rep3_ERCC-Mix1/gene_read_counts_table.tsv
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/HBR_Rep1_ERCC-Mix2/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > HBR_Rep1_ERCC-Mix2/gene_read_counts_table.tsv
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/HBR_Rep2_ERCC-Mix2/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > HBR_Rep2_ERCC-Mix2/gene_read_counts_table.tsv
htseq-count --format bam --order pos --mode intersection-strict --stranded reverse --minaqual 1 --type exon --idattr gene_id $RNA_HOME/alignments/tophat/HBR_Rep3_ERCC-Mix2/accepted_hits.bam $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf > HBR_Rep3_ERCC-Mix2/gene_read_counts_table.tsv
Merge results files into a single matrix for use in edgeR:
join UHR_Rep1_ERCC-Mix1/gene_read_counts_table.tsv UHR_Rep2_ERCC-Mix1/gene_read_counts_table.tsv | join - UHR_Rep3_ERCC-Mix1/gene_read_counts_table.tsv | join - HBR_Rep1_ERCC-Mix2/gene_read_counts_table.tsv | join - HBR_Rep2_ERCC-Mix2/gene_read_counts_table.tsv | join - HBR_Rep3_ERCC-Mix2/gene_read_counts_table.tsv > gene_read_counts_table_all.tsv
Based on the above read counts, plot the linearity of the ERCC spike-in read counts versus the known concentration of the ERCC spike-in Mix:
cd $RNA_HOME/expression/tophat_counts
wget http://genome.wustl.edu/pub/rnaseq/data/brain_vs_uhr_w_ercc/ERCC/ERCC_Controls_Analysis.txt
cat ERCC_Controls_Analysis.txt
wget https://raw.githubusercontent.com/griffithlab/rnaseq_tutorial/master/scripts/Tutorial_Module4_ERCC_expression.pl
chmod +x Tutorial_Module4_ERCC_expression.pl
./Tutorial_Module4_ERCC_expression.pl
cat $RNA_HOME/expression/tophat_counts/ercc_read_counts.tsv
wget https://raw.githubusercontent.com/griffithlab/rnaseq_tutorial/master/scripts/Tutorial_Module4_ERCC_expression.R
chmod +x Tutorial_Module4_ERCC_expression.R
./Tutorial_Module4_ERCC_expression.R ercc_read_counts.tsv
To view the resulting figure, navigate to the below URL replacing YOUR_IP_ADDRESS with your amazon instance IP address:
- http://YOUR_IP_ADDRESS/workspace/rnaseq/expression/tophat_counts/Tutorial_Module4_ERCC_expression.pdf
| Previous Section | This Section | Next Section | |:-------------------------------------:|:---------------------------:|:--------------------------------------------------------:| | Alignment QC | Expression | Differential Expression |