Alignment
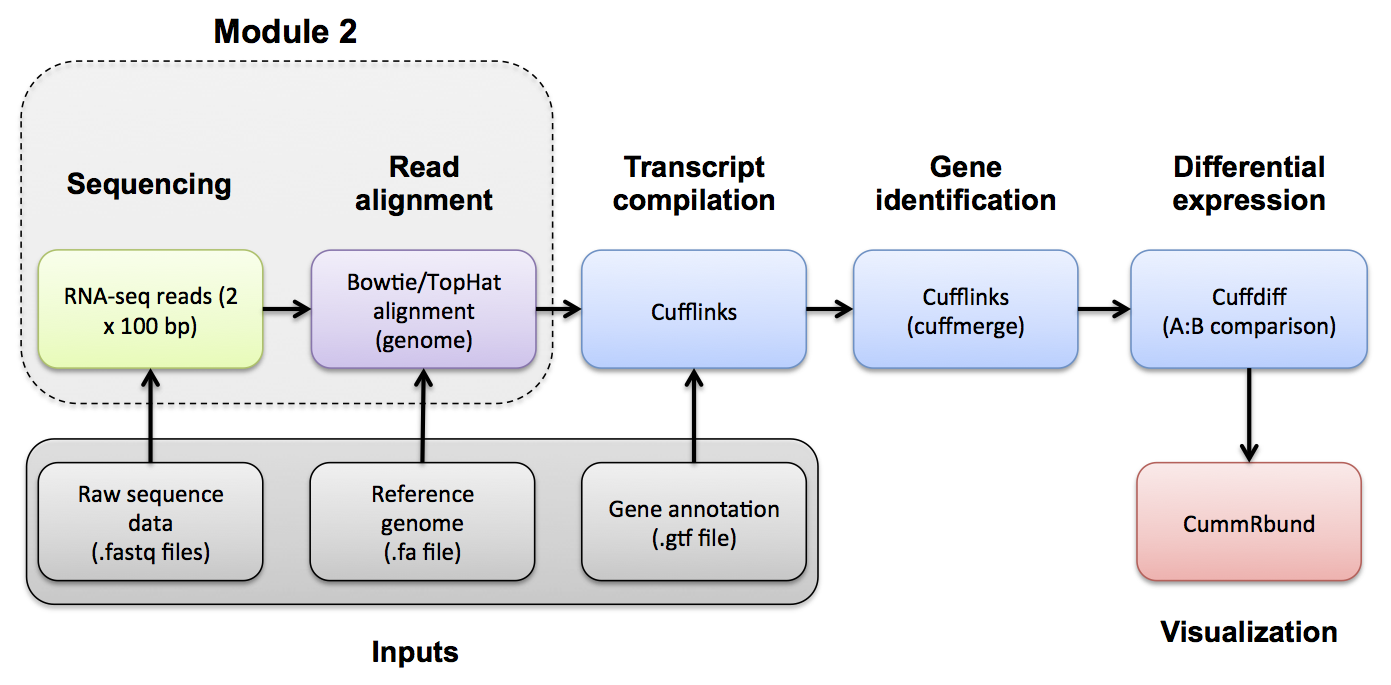
#2-ii. Alignment Use Bowtie2/Tophat2 to align all pairs of read files to the genome. The output of this step will be a SAM/BAM file for each data set.
Refer to TopHat manual and tutorial for a more detailed explanation:
TopHat2 basic usage:
#tophat2 [options] <bowtie_index> <lane1_reads1[,lane2_reads1,...]> <lane1_reads2[,lane2_reads2,...]>
Extra options specified below:
- '-p 8' tells TopHat to use eight CPUs for bowtie alignments
- '-r 60' tells TopHat the expected inner distance between the reads of a pair. [fragment size - (2 x read length)]. 260 - (2 x 100) = 60
- '--library-type fr-firststrand' since the TruSeq strand-specific library was used to make these libraries
- '-o' tells TopHat to write the output to a particular directory (one per sample)
- '--rg-id' specifies a read group ID
- '--rg-sample' specified a read group sample ID. This together with rg-id will allow you to determine which reads came from which library in the merged bam later on
- '-G ' supplies a list of known transcript models. These will be used to help TopHat measure known exon-exon connections (novel connections will still be predicted)
- Note that the '-G' option for TopHat has a different meaning than the '-G' option of Cufflinks that we will use in step 9 later
- '--transcriptome-index' TopHat will align to both the transcriptome and genome and figure out the 'best' alignments for you.
- In order to perform alignments to the transcriptome, an index must be created as we did for the genome.
- This parameter tells TopHat where to store it and allows it to be reused in multiple TopHat runs.
##TopHat alignment
cd $RNA_HOME/
export RNA_DATA_DIR=$RNA_HOME/data/
echo $RNA_DATA_DIR
mkdir -p alignments/tophat/trans_idx
cd alignments/tophat
export TRANS_IDX_DIR=$RNA_HOME/alignments/tophat/trans_idx/
echo $TRANS_IDX_DIR
In our tests, each sample took ~1-1.5 minutes to align
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=UHR_Rep1 --rg-sample=UHR_Rep1_ERCC-Mix1 -o UHR_Rep1_ERCC-Mix1 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=UHR_Rep2 --rg-sample=UHR_Rep2_ERCC-Mix1 -o UHR_Rep2_ERCC-Mix1 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=UHR_Rep3 --rg-sample=UHR_Rep3_ERCC-Mix1 -o UHR_Rep3_ERCC-Mix1 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=HBR_Rep1 --rg-sample=HBR_Rep1_ERCC-Mix2 -o HBR_Rep1_ERCC-Mix2 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=HBR_Rep2 --rg-sample=HBR_Rep2_ERCC-Mix2 -o HBR_Rep2_ERCC-Mix2 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz
tophat2 -p 8 -r 60 --library-type fr-firststrand --rg-id=HBR_Rep3 --rg-sample=HBR_Rep3_ERCC-Mix2 -o HBR_Rep3_ERCC-Mix2 -G $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf --transcriptome-index $TRANS_IDX_DIR/ENSG_Genes $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/chr22_ERCC92 $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz
Note: in the above alignments, we are treating each library as an independent data set. If you had multiple lanes of data for a single library, you would want to align them all together in one TopHat command Similarly you might combine technical replicates into a single alignment run (perhaps after examining them and removing outliers...) To combine multiple lanes, you would provide all the read1 files as a comma separated list, followed by a space, and then all read2 files as a comma separated list (where both lists have the same order): You can also use samtools merge to combine bam files after alignment. This is the approach we will take.
###TopHat Alignment Summary TopHat generates a summary of the alignments in a text file next to the aligned BAM file. The below command will print the summary for each alignment to the terminal.
cat */align_summary.txt
###OPTIONAL ALTERNATIVE - STAR alignment Perform alignments with STAR. STAR alignment results can be used for Cufflinks analysis or other further RNA-seq analysis. Some further optional parameters might be needed though (see STAR manual: 8.2.3: XS SAM strand attribute for Cufflinks/Cuffdiff).
cd $RNA_HOME/
mkdir -p alignments/star
cd alignments/star
mkdir UHR_Rep1 UHR_Rep2 UHR_Rep3 HBR_Rep1 HBR_Rep2 HBR_Rep3
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix UHR_Rep1/
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix UHR_Rep2/
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix UHR_Rep3/
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix HBR_Rep1/
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix HBR_Rep2/
STAR --genomeDir $RNA_HOME/refs/hg19/star/chr22_ERCC92 --readFilesIn $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz --runThreadN 8 --readFilesCommand zcat --outFileNamePrefix HBR_Rep3/
Convert STAR sam files to bam files (required for cufflinks)
samtools view -b -S UHR_Rep1/Aligned.out.sam > UHR_Rep1/Aligned.out.bam
samtools view -b -S UHR_Rep2/Aligned.out.sam > UHR_Rep2/Aligned.out.bam
samtools view -b -S UHR_Rep3/Aligned.out.sam > UHR_Rep3/Aligned.out.bam
samtools view -b -S HBR_Rep1/Aligned.out.sam > HBR_Rep1/Aligned.out.bam
samtools view -b -S HBR_Rep2/Aligned.out.sam > HBR_Rep2/Aligned.out.bam
samtools view -b -S HBR_Rep3/Aligned.out.sam > HBR_Rep3/Aligned.out.bam
Now sort STAR bam files (also required for cufflinks)
samtools sort UHR_Rep1/Aligned.out.bam -o UHR_Rep1/Aligned.out.sorted.bam
samtools sort UHR_Rep2/Aligned.out.bam -o UHR_Rep2/Aligned.out.sorted.bam
samtools sort UHR_Rep3/Aligned.out.bam -o UHR_Rep3/Aligned.out.sorted.bam
samtools sort HBR_Rep1/Aligned.out.bam -o HBR_Rep1/Aligned.out.sorted.bam
samtools sort HBR_Rep2/Aligned.out.bam -o HBR_Rep2/Aligned.out.sorted.bam
samtools sort HBR_Rep3/Aligned.out.bam -o HBR_Rep3/Aligned.out.sorted.bam
###Optional Alternative - HISAT2 alignment Perform alignments with HISAT2. HISAT2 uses a graph-based alignment and has succeeded HISAT and TOPHAT2.
cd $RNA_HOME/
mkdir -p alignments/hisat2
cd alignments/hisat2
mkdir UHR_Rep1 UHR_Rep2 UHR_Rep3 HBR_Rep1 HBR_Rep2 HBR_Rep3
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep1/Aligned.out.sam
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep2/Aligned.out.sam
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep3/Aligned.out.sam
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep1/Aligned.out.sam
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep2/Aligned.out.sam
hisat2 -p 8 -x $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/chr22_ERCC92 --known-splicesite-infile $RNA_HOME/refs/hg19/hisat2/chr22_ERCC92/splicesites.txt --dta-cufflinks --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep3/Aligned.out.sam
Convert HISAT2 sam files to bam files (required for cufflinks)
samtools view -b -S UHR_Rep1/Aligned.out.sam > UHR_Rep1/Aligned.out.bam
samtools view -b -S UHR_Rep2/Aligned.out.sam > UHR_Rep2/Aligned.out.bam
samtools view -b -S UHR_Rep3/Aligned.out.sam > UHR_Rep3/Aligned.out.bam
samtools view -b -S HBR_Rep1/Aligned.out.sam > HBR_Rep1/Aligned.out.bam
samtools view -b -S HBR_Rep2/Aligned.out.sam > HBR_Rep2/Aligned.out.bam
samtools view -b -S HBR_Rep3/Aligned.out.sam > HBR_Rep3/Aligned.out.bam
Now sort the HISAT2 bam files (also required for cufflinks)
samtools sort UHR_Rep1/Aligned.out.bam -o UHR_Rep1/Aligned.out.sorted.bam
samtools sort UHR_Rep2/Aligned.out.bam -o UHR_Rep2/Aligned.out.sorted.bam
samtools sort UHR_Rep3/Aligned.out.bam -o UHR_Rep3/Aligned.out.sorted.bam
samtools sort HBR_Rep1/Aligned.out.bam -o HBR_Rep1/Aligned.out.sorted.bam
samtools sort HBR_Rep2/Aligned.out.bam -o HBR_Rep2/Aligned.out.sorted.bam
samtools sort HBR_Rep3/Aligned.out.bam -o HBR_Rep3/Aligned.out.sorted.bam
##Merge TopHat BAM files Make one glorious BAM combining all UHR data and another for all HBR data. Note: This could be done in several ways such as 'samtools merge', 'bamtools merge', or using picard-tools (see below). We chose the third method because it did the best job at merging the bam header information.
UHR
cd $RNA_HOME/alignments/tophat
mkdir UHR_ERCC-Mix1_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=UHR_ERCC-Mix1_ALL/accepted_hits.bam INPUT=UHR_Rep1_ERCC-Mix1/accepted_hits.bam INPUT=UHR_Rep2_ERCC-Mix1/accepted_hits.bam INPUT=UHR_Rep3_ERCC-Mix1/accepted_hits.bam
HBR
cd $RNA_HOME/alignments/tophat
mkdir HBR_ERCC-Mix2_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=HBR_ERCC-Mix2_ALL/accepted_hits.bam INPUT=HBR_Rep1_ERCC-Mix2/accepted_hits.bam INPUT=HBR_Rep2_ERCC-Mix2/accepted_hits.bam INPUT=HBR_Rep3_ERCC-Mix2/accepted_hits.bam
Count the alignment (BAM) files to make sure all were created successfully (you should have 8 total)
ls -l */accepted_hits.bam | wc -l
ls -l */accepted_hits.bam
###OPTIONAL ALTERNATIVE - Merge STAR bam files Create comparable files for the STAR alignments by merging individual bam files generated by STAR.
cd $RNA_HOME/alignments/star
mkdir UHR_ERCC-Mix1_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=UHR_ERCC-Mix1_ALL/Aligned.out.sorted.bam INPUT=UHR_Rep1/Aligned.out.sorted.bam INPUT=UHR_Rep2/Aligned.out.sorted.bam INPUT=UHR_Rep3/Aligned.out.sorted.bam
mkdir HBR_ERCC-Mix2_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=HBR_ERCC-Mix2_ALL/Aligned.out.sorted.bam INPUT=HBR_Rep1/Aligned.out.sorted.bam INPUT=HBR_Rep2/Aligned.out.sorted.bam INPUT=HBR_Rep3/Aligned.out.sorted.bam
###OPTIONAL ALTERNATIVE - merge HISAT2 bam files Create comparable files for the HISAT2 alignments by merging individual bam files generated by HISAT2
cd $RNA_HOME/alignments/hisat2
mkdir UHR_ERCC-Mix1_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=UHR_ERCC-Mix1_ALL/Aligned.out.sorted.bam INPUT=UHR_Rep1/Aligned.out.sorted.bam INPUT=UHR_Rep2/Aligned.out.sorted.bam INPUT=UHR_Rep3/Aligned.out.sorted.bam
mkdir HBR_ERCC-Mix2_ALL
java -Xmx2g -jar $RNA_HOME/tools/picard-tools-1.140/picard.jar MergeSamFiles OUTPUT=HBR_ERCC-Mix2_ALL/Aligned.out.sorted.bam INPUT=HBR_Rep1/Aligned.out.sorted.bam INPUT=HBR_Rep2/Aligned.out.sorted.bam INPUT=HBR_Rep3/Aligned.out.sorted.bam
##PRACTICAL EXERCISE 5
Assignment: Perform some alignments on an additional pair of read data sets. Align the reads using the skills you learned above. Try using Tophat or STAR aligners.
- Hint: Do this analysis on the additional data and in the separate working directory called ‘practice’ that you created in Practical Exercise 2.
Solution: When you are ready you can check your approach against the Solutions
| Previous Section | This Section | Next Section | |:---------------------------------:|:--------------------------:|:-----------------------------:| | Adapter Trim | Alignment | IGV |