Home
Announcements: Now provides simplified integration for existing STAR-Fusion users.
CTAT-Mutations Pipeline is a variant calling pipeline focussed on detecting variants from RNA sequencing (RNA-seq) data. It integrates GATK Best Practices along with downstream steps to annotate and filter variants, and to additionally prioritize variants that may be relevant to cancer biology such as likely somatic mutations. Our variant annotation includes leveraging the RADAR and RediPortal databases for identifying likely RNA-editing events, dbSNP and gnomAD for annotating common variants, and COSMIC to highlight known cancer mutations. Finally, CRAVAT is leveraged to annotate and prioritize variants according to likely biological impact and relevance to cancer.
The CTAT Mutations pipeline is one of the components of the Trinity Cancer Transcriptome Analysis Toolkit (CTAT), complementing other functionality that leverages RNA-Seq data for characterizing cancer transcriptomes, including identification of fusion transcripts, copy number variations from tumor single cell transcriptomes, among other capabilities.
Our CTAT-Mutation pipeline aims to make variant discovery from rna-seq data as easy as possible, requiring only the RNA-seq reads as input, and generating summary reports and visualizations to help guide you to the most meaningful findings.
The following flowchart is a simplified visualization of the steps performed by the CTAT-Mutation Pipeline.
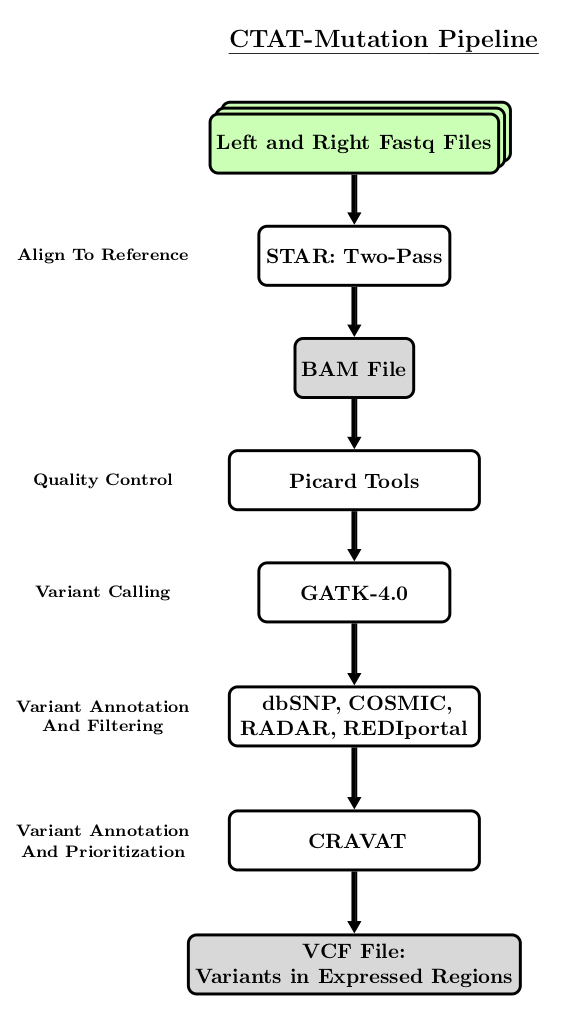
The CTAT-Mutations pipeline requires the CTAT-Mutations software and companion genomic data resources. See our instructions for installing CTAT-Mutations for details.
Once the CTAT-Mutation Pipeline has successfully been installed along with the obligatory CTAT Genome Library, CTAT-Mutation Pipeline can be ran using the following command, only requiring the input reads.
python /path/to/ctat_mutations \
--left : Path to the location of the left (ie. /1) paired end RNA-Seq Fastq file.
--right : Path to the location of the right (ie, /2) paired end RNA-Seq Fastq file.
--sample_id : The sample id
--outputdir : Name to be given to the directory in which CTAT-Mutation outputs will
be placed.
As inputs, CTAT-Mutation requires RNA-Seq reads in the form of a right and left paired-end FASTQ files, along with an output directory name where the pipeline products will be stored.
A small sample data set comes with ctat_mutations and is available for testing purposes. The pipeline can be ran on the sample data set by running the following command:
python /path/to/ctat_mutations \
--left reads_1.fastq.gz \
--right reads_2.fastq.gz \
--outputdir varcalling.outdir \
--sample_id test
See our more detailed walk through tutorial leveraging these data.
By default, gradient boosting is applied to augment prediction accuracy. You can set the flag --boosting_method to none to apply hard filters instead of boosting. Additional machine learning approaches are incorporated for reducing false positive calls, including gradient boosting, stochastic gradient boosting, adaptive boosting, and random forests. Hard-filtering and boosting methods are mutually exclusive. You can first run with the boosting enabled and then run again with hard filtering, and the second run will reuse preexisting outputs from the earlier execution where possible, speeding up the process. See Output section below for more details.
The output from the CTAT-Mutations pipeline includes variant vcf files, summary tab-delimited reports, and interactive visualizations.
The primary outputs include variants.HC_init.wAnnot.vcf.gz, containing HaplotypeCaller variant calls fully annotated. If boosting or hard-cutoffs are applied for filtering, additional corresponding vcfs are provided containing the unfiltered variant calls. A cancer.vcf and corresponding simpler summary cancer.tab file, are provided that contain a set of prioritized cancer-relevant variants detected in the sample. The cancer.vcf (VCF version 4.0) records the genetic variations, their locations, and additional annotation information. The cancer.tab is a tab-delimited file that contains the same variant information in a user-friendly format. There are additional outputs that are generated by the different stages of the CTAT-Mutations pipeline, as others are likely to be of interest as well for exploring RNA-editing or common variants. Documentation is provided for all such output files and formats.
You will also find an html page output named "igvjs_viewer.html" (based on igv-reports ), which allows for dynamic navigation of the identified cancer variants and the read evidence supporting their identification. This file can be simply opened in your web browser. An example view is shown below.

More info for exploring the variant visualization framework is available here.
The igv-reports based html report derives from a collaboration with James Robinson.
CTAT-Mutations is available for running on Terra and easy to use - select the Hg19 or Hg38 based workflow from the web interface, specify your RNA-seq fastq inputs, and click 'go'.
Contact us on our google group https://groups.google.com/forum/#!forum/trinity_ctat_users
We aim to be responsive with user support. You will be responded to within hours time, generally (not days or weeks).
CTAT-Mutations is supported as part of the Trinity CTAT Project, funded by the National Cancer Institute Informatics Technology for Cancer Research