single cell variant calling
Our current approach for detecting variants and mutations in single cells involves running CTAT-Mutations on all reads, identifying variants, and then identifying the cells expressing reference or alternate allelic variants.
Before running single cell RNA-seq through CTAT-Mutations, the names of the reads should be encoded with cell barcode and UMI information in the following format:
cellbarcode^UMI^read_name
If you have 10xGenomics reads in a ubam format, you can convert to fastq format with the above read name encoding using this script: 10x_ubam_to_fastq.py
Then, run CTAT-Mutations using this fastq file as input along with the command-line flag '--is_single_cells'
When run with parameter '--is_single_cells', an additional output file will be generated: '${sample_name}.annot_pass_reads.vcf.sc_reads.gz' with the following format:
chr_pos_variant num_reads_with_variant reads_with_variant num_ref_matching_reads ref_matching_reads
chr1:14436:G:A 25 TTCGAAGTCACGACTA^GACCAGGGTTTCCCACCAAC^molecule/84621990,ATTATCCCAATCACAC^TTTCGGCCAATTTCTTATAT^molecule/18857150,... 271 GTAGTCACAGAAGCAC^TTGTCCTGCGTTTCTTATAT^molecule/63537222,ATCATCTTCCAGATCA^TTTACATGCCTTTCTTATAT^molecule/16809660,...
...
which includes, in tab-delimited format:
- the variant found, formatted as: chromosome:position:REF:ALT
- number of reads that contain the variant
- identity of the reads (comma-delimited) that contain the variant
- number of reads that match the reference allele
- identification of the reads that contain the reference allele
This file can be subsequently processed to extract a summary of counts of UMIs that support the REF and ALT alleles for every cell like so:
${CTAT_MUTATIONS_INSTALLDIR}/src/SingleCells/variant_cell_UMI_count_report.py \
--vcf_sc_reads_tsv ${sample_name}.annot_pass_reads.vcf.sc_reads.gz \
--output ${sample_name}.annot_pass_reads.vcf.sc_reads.variant_to_cell
chr_pos_variant cell_barcode num_reads_w_variant num_ref_matching_reads
chr10:100000235:C:T ACGAGCCCACTATCTT 1.0 0.0
chr10:100000235:C:T ATCTACTGTCTGATCA 1.0 0.0
chr10:100000235:C:T CAAGTTGTCACCACCT 1.0 0.0
chr10:100000235:C:T CATTCGCCACTTACGA 1.0 0.0
chr10:100000235:C:T CCATGTCTCCTAGTGA 1.0 0.0
chr10:100000235:C:T GCAGCCACATCGACGC 1.0 0.0
chr10:100000235:C:T GCAGTTATCCCTCTTT 1.0 0.0
chr10:100000235:C:T GGCGTGTCACGGACAA 1.0 0.0
chr10:100000235:C:T GTACGTAAGAGGTACC 1.0 0.0
chr10:100000235:C:T GTGCATATCCAGAAGG 1.0 0.0
chr10:100000235:C:T GTTCGGGTCGGTGTTA 1.0 0.0
chr10:100000235:C:T TCACGAATCAATAAGG 1.0 0.0
chr10:100000235:C:T TGAAAGATCAGGATCT 1.0 0.0
chr10:100000235:C:T TGACTAGTCATTCACT 1.0 0.0
chr10:100000235:C:T TGAGCCGAGTTGTAGA 1.0 0.0
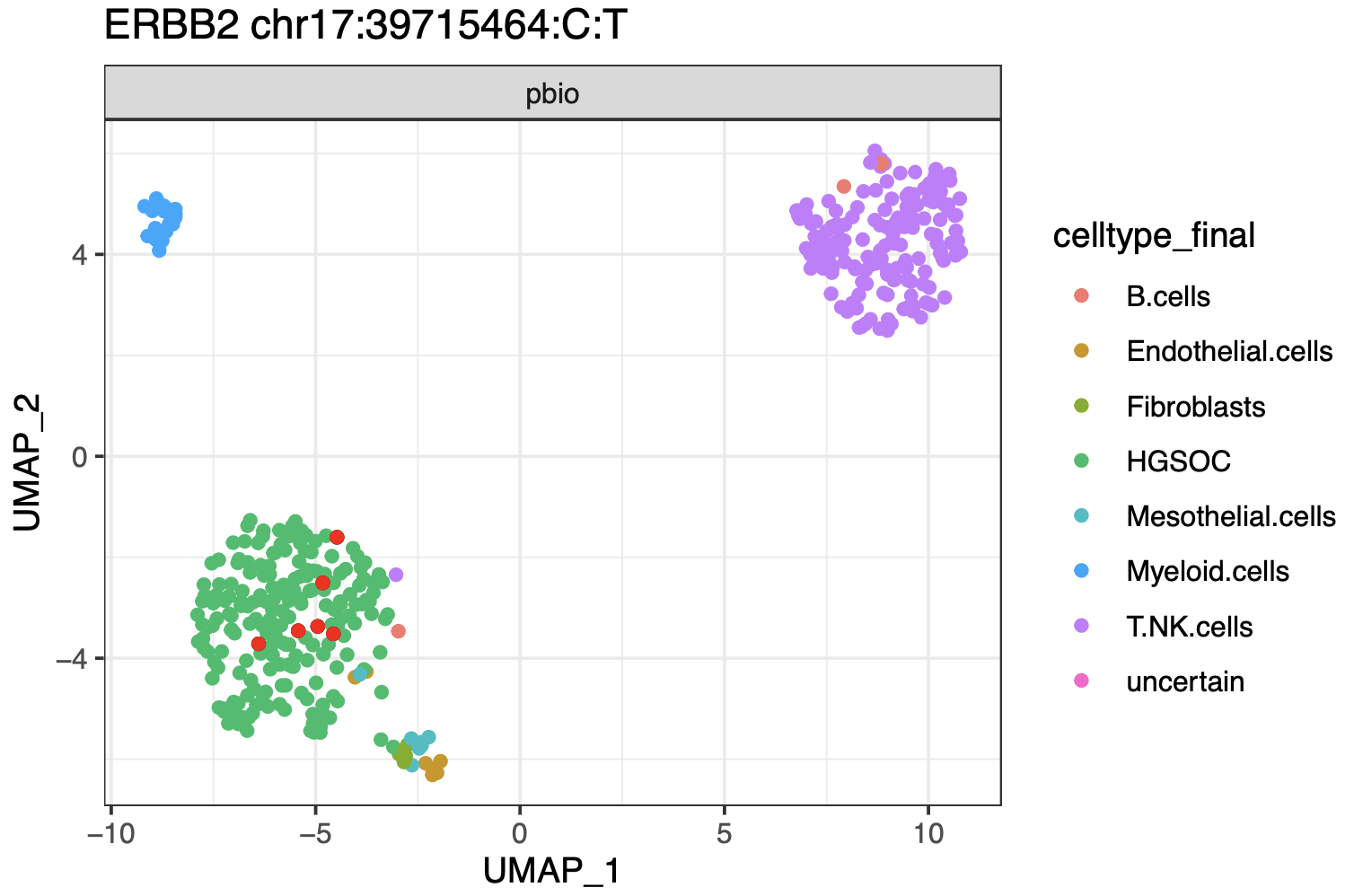
From this output, you can extract for every variant the cells that express the REF and/or the ALT allele, and explore these further using single cell analysis techniques - such as painting variant containing cells in a UMAP plot, or identifying those variants that show up in tumor cells as opposed to normal cells and likely represent somatic mutations: