![]()
![]()
![]()
Please cite:
Serizay J, Matthey-Doret C, Bignaud A, Baudry L, Koszul R (2024). “Orchestrating chromosome conformation capture analysis with Bioconductor.” Nature Communications, 15, 1-9. doi:10.1038/s41467-024-44761-x.
HiContacts provides tools to investigate (m)cool matrices imported in R by HiCExperiment.
It leverages the HiCExperiment class of objects, built on pre-existing Bioconductor objects, namely InteractionSet, GInterations and ContactMatrix (Lun, Perry & Ing-Simmons, F1000Research 2016), and provides analytical and visualization tools to investigate contact maps.
HiContacts is available in Bioconductor. To install the current release, use:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("HiContacts")To install the most recent version of HiContacts, you can use:
install.packages("devtools")
devtools::install_github("js2264/HiContacts")
library(HiContacts)If you are using HiContacts in your research, please cite:
Serizay J (2022). HiContacts: HiContacts: R interface to cool files. R package version 1.1.0 https://github.com/js2264/HiContacts.
HiContacts includes a introduction vignette where its usage is
illustrated. To access the vignette, please use:
vignette('HiContacts')mcool_file <- HiContactsData::HiContactsData('yeast_wt', format = 'mcool')
range <- 'I:20000-80000' # range of interest
availableResolutions(mcool_file)
hic <- HiCExperiment::import(mcool_file, format = 'mcool', focus = range, resolution = 1000)
hicplotMatrix(hic, use.scores = 'count')
plotMatrix(hic, use.scores = 'balanced', limits = c(-4, -1))
plotMatrix(hic, use.scores = 'balanced', limits = c(-4, -1), maxDistance = 100000)library(rtracklayer)
mcool_file <- HiContactsData::HiContactsData('yeast_wt', format = 'mcool')
hic <- import(mcool_file, format = 'mcool', focus = 'IV')
loops <- system.file("extdata", 'S288C-loops.bedpe', package = 'HiContacts') |>
import() |>
InteractionSet::makeGInteractionsFromGRangesPairs()
borders <- system.file("extdata", 'S288C-borders.bed', package = 'HiContacts') |>
import()
p <- plotMatrix(
hic, loops = loops, borders = borders,
limits = c(-4, -1), dpi = 120
)contacts <- contacts_yeast()
contacts <- zoom(contacts, resolution = 2000)
aggr_centros <- aggregate(contacts, targets = topologicalFeatures(contacts, 'centromeres'))
plotMatrix(aggr_centros, use.scores = 'detrended', limits = c(-1, 1), scale = 'linear')microC_mcool <- fourDNData::fourDNData('4DNES14CNC1I', 'mcool')
hic <- import(microC_mcool, format = 'mcool', resolution = 10000000)
genome <- BSgenome.Mmusculus.UCSC.mm10::BSgenome.Mmusculus.UCSC.mm10
# - Get compartments
hic <- getCompartments(
hic, resolution = 100000, genome = genome, chromosomes = c('chr17', 'chr19')
)
# - Export compartments as bigwig and bed files
export(IRanges::coverage(metadata(hic)$eigens, weight = 'eigen'), 'microC_compartments.bw')
export(
topologicalFeatures(hic, 'compartments')[topologicalFeatures(hic, 'compartments')$compartment == 'A'],
'microC_A-compartments.bed'
)
export(
topologicalFeatures(hic, 'compartments')[topologicalFeatures(hic, 'compartments')$compartment == 'B'],
'microC_B-compartments.bed'
)
# - Generate saddle plot
plotSaddle(hic)# - Compute insulation score
hic <- refocus(hic, 'chr19:1-30000000') |>
zoom(resolution = 10000) |>
getDiamondInsulation(window_size = 100000) |>
getBorders()
# - Export insulation as bigwig track and borders as bed file
export(IRanges::coverage(metadata(hic)$insulation, weight = 'insulation'), 'microC_insulation.bw')
export(topologicalFeatures(hic, 'borders'), 'microC_borders.bed')hic <- import(CoolFile(
mcool_file,
pairs = HiContactsData::HiContactsData('yeast_wt', format = 'pairs.gz')
))
ps <- distanceLaw(hic)
plotPs(ps, ggplot2::aes(x = binned_distance, y = norm_p))hic <- import(CoolFile(mcool_file))
v4C <- virtual4C(hic, viewpoint = GRanges('V:150000-170000'))
plot4C(v4C)hic <- import(CoolFile(mcool_file))
cisTransRatio(hic)HiCool is integrated within the HiCExperiment ecosystem in Bioconductor.
Read more about the HiCExperiment class and handling Hi-C data in R
here.
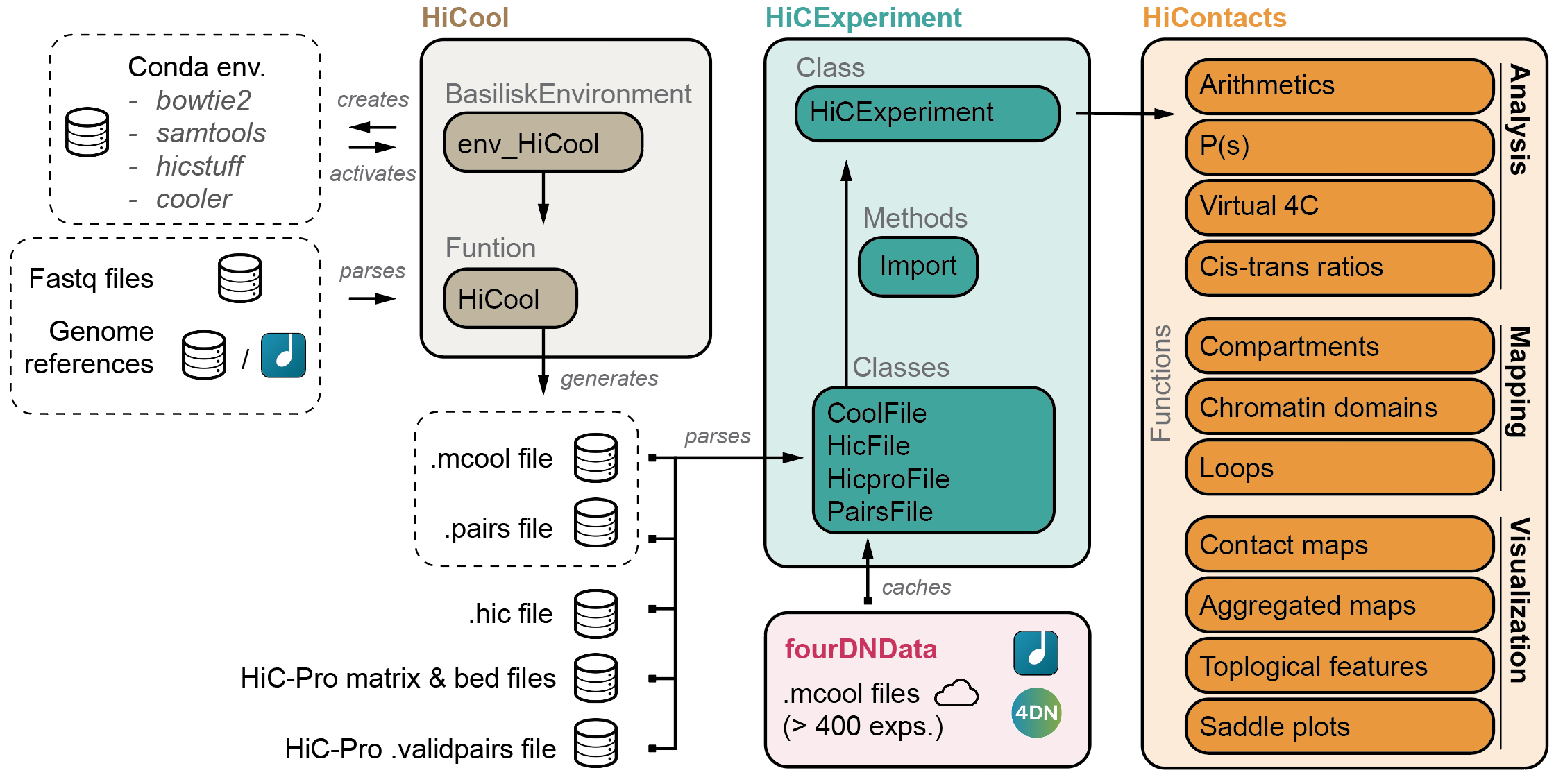
- HiCExperiment: Parsing Hi-C files in R
- HiCool: End-to-end integrated workflow to process fastq files into .cool and .pairs files
- HiContacts: Investigating Hi-C results in R
- HiContactsData: Data companion package
- fourDNData: Gateway package to 4DN-hosted Hi-C experiments