![]()
![]()
![]()
Please cite:
Serizay J, Matthey-Doret C, Bignaud A, Baudry L, Koszul R (2024). “Orchestrating chromosome conformation capture analysis with Bioconductor.” Nature Communications, 15, 1-9. doi:10.1038/s41467-024-44761-x.
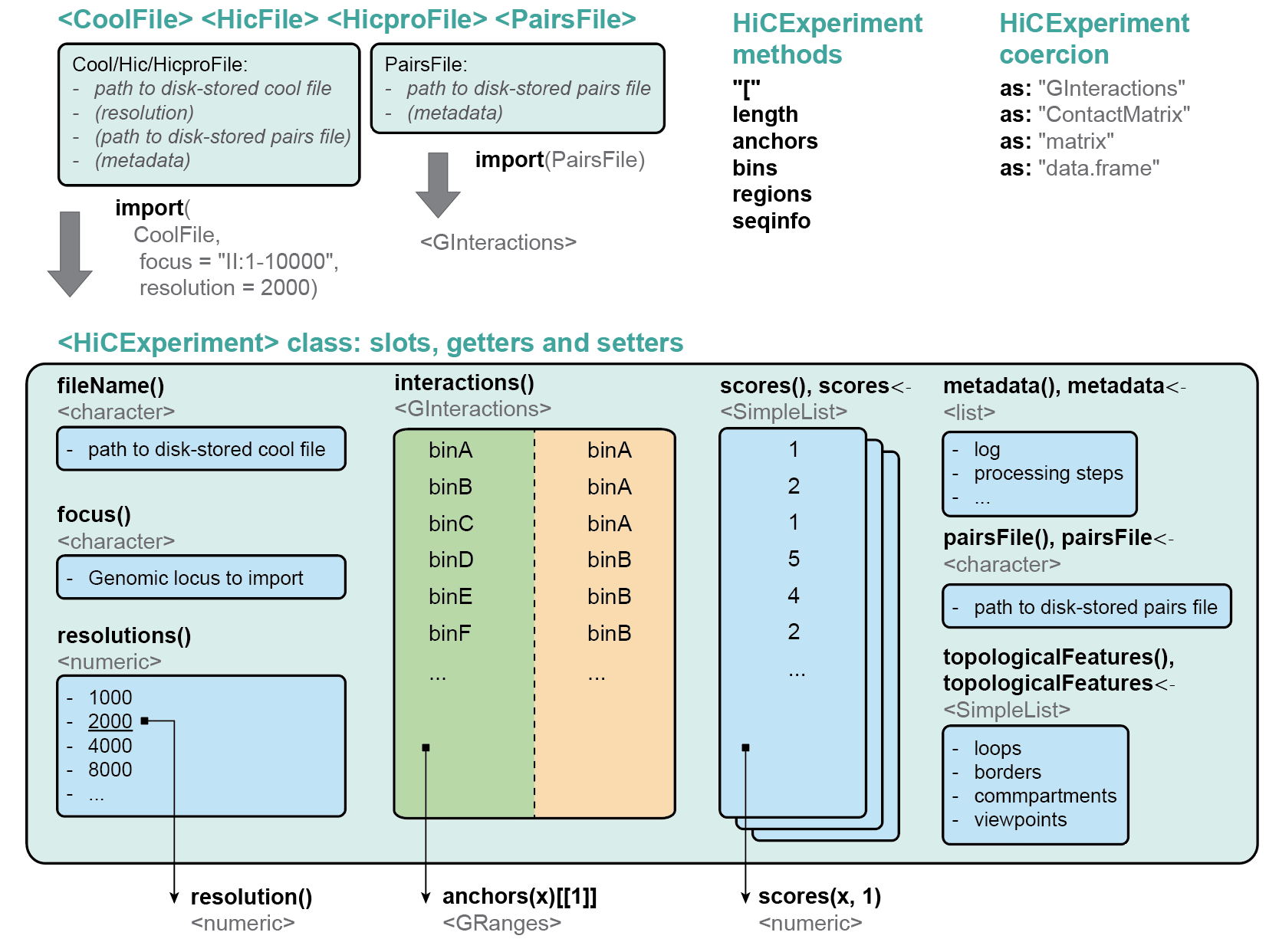
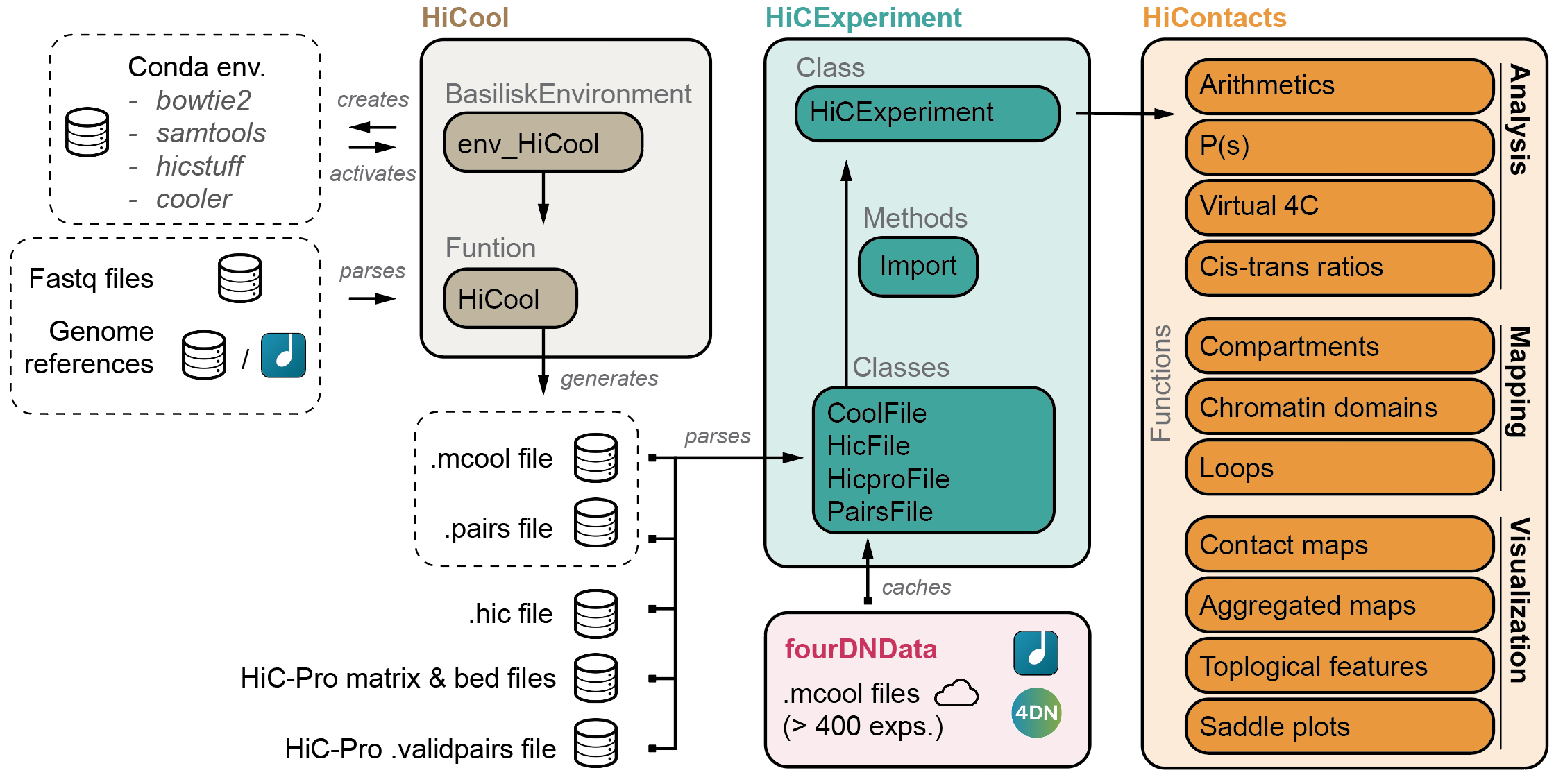
The HiCExperiment package provides a unified data structure to import the three main Hi-C matrix file formats (.(m)cool, .hic and HiC-Pro matrices) in R and performs common array operations on them.
The HiCExperiment class wraps an (indexed) matrix-like object (i.e. on-disk .(m)cool, .hic or HiC-Pro matrices). For indexed matrices (i.e. .(m)cool and .hic files), HiCExperiment allows one to specfically parse subsets of the contact matrix corresponding to genomic loci of interest, without having to load the entire object in memory.
The HiCExperiment package also provides methods to import pairs files generated by pairtools/cooler workflow, by HiC-Pro pipeline, or any type of tabular pairs format (by indicating the columns containing chr1, start1, strand1, chr2, start2, strand2 information).
HiCExperiment S4 class is built on pre-existing Bioconductor classes, namely BiocFile and
GInteractions (Lun, Perry & Ing-Simmons, F1000Research 2016`), and leverages them to
point to on-disk Hi-C matrix files and dynamically parse them into R.
Several other packages rely on the HiCExperiment class to provide a rich ecosystem when interacting with Hi-C data.
HiCExperiment is an R/Bioconductor package. As such, it can be installed with:
BiocManager::install("HiCExperiment")cool_file <- CoolFile(HiContactsData::HiContactsData('yeast_wt', format = 'cool'))
import(cool_file, focus = "II:10000-100000")## `HiCExperiment` object with 3,454 interactions over 90 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/36d548fb47bf_7751"
## focus: "II:10,000-100,000"
## resolutions(1): 1000
## current resolution: 1000
## interactions: 3454
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
mcool_file <- CoolFile(HiContactsData::HiContactsData('yeast_wt', format = 'mcool'))
import(mcool_file, focus = "II:10000-100000", resolution = 2000)## `HiCExperiment` object with 1,004 interactions over 45 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/36d590c5583_7752"
## focus: "II:10,000-100,000"
## resolutions(5): 1000 2000 4000 8000 16000
## current resolution: 2000
## interactions: 1004
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
hic_file <- HicFile(HiContactsData::HiContactsData('yeast_wt', format = 'hic'))
import(hic_file, focus = "II:10000-100000", resolution = 4000)## `HiCExperiment` object with 276 interactions over 23 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/7fa45373d163_7836"
## focus: "II:10,000-100,000"
## resolutions(5): 1000 2000 4000 8000 16000
## current resolution: 4000
## interactions: 276
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
hicpro_file <- HicproFile(
HiContactsData::HiContactsData('yeast_wt', format = 'hicpro_matrix'),
bed = HiContactsData::HiContactsData('yeast_wt', format = 'hicpro_bed')
)
import(hicpro_file)## `HiCExperiment` object with 2,686,250 interactions over 11,805 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/29210052806_7837"
## focus: "whole genome"
## resolutions(1): 1000
## current resolution: 1000
## interactions: 2686250
## scores(1): counts
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(1): regions
.pairsfiles (e.g. frompairtoolsorcooler):
pairs_file <- PairsFile(HiContactsData('yeast_wt', format = 'pairs.gz'))
import(pairs_file)## GInteractions object with 471364 interactions and 4 metadata columns:
## seqnames1 ranges1 seqnames2 ranges2 | counts frag1 frag2 distance
## <Rle> <IRanges> <Rle> <IRanges> | <integer> <numeric> <numeric> <numeric>
## [1] II 105 --- II 48548 | 1 1358 1681 48443
## [2] II 113 --- II 45003 | 1 1358 1658 44890
## [3] II 119 --- II 687251 | 1 1358 5550 687132
## [4] II 160 --- II 26124 | 1 1358 1510 25964
## [5] II 169 --- II 39052 | 1 1358 1613 38883
## ... ... ... ... ... ... . ... ... ... ...
## [471360] II 808605 --- II 809683 | 1 6316 6320 1078
## [471361] II 808609 --- II 809917 | 1 6316 6324 1308
## [471362] II 808617 --- II 809506 | 1 6316 6319 889
## [471363] II 809447 --- II 809685 | 1 6319 6321 238
## [471364] II 809472 --- II 809675 | 1 6319 6320 203
## -------
## regions: 549331 ranges and 0 metadata columns
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
.validPairsfiles (e.g. from HiC-Pro pipeline):
hicpro_pairs_file <- PairsFile(HiContactsData('yeast_wt', format = 'hicpro_pairs'))
import(hicpro_pairs_file, nrows = 100)## GInteractions object with 100 interactions and 4 metadata columns:
## seqnames1 ranges1 seqnames2 ranges2 | counts frag1 frag2 distance
## <Rle> <IRanges> <Rle> <IRanges> | <integer> <numeric> <character> <numeric>
## [1] I 33 --- I 620 | 1 414 HIC_I_1 587
## [2] I 35 --- III 301620 | 1 336 HIC_I_1 NA
## [3] I 41 --- I 68853 | 1 352 HIC_I_1 68812
## [4] I 49 --- I 3233 | 1 311 HIC_I_1 3184
## [5] I 51 --- VIII 197898 | 1 397 HIC_I_1 NA
## ... ... ... ... ... ... . ... ... ... ...
## [96] I 138 --- VIII 326284 | 1 251 HIC_I_1 NA
## [97] I 141 --- I 2466 | 1 231 HIC_I_1 2325
## [98] I 142 --- I 2219 | 1 278 HIC_I_1 2077
## [99] I 142 --- XI 222517 | 1 270 HIC_I_1 NA
## [100] I 142 --- XV 441757 | 1 280 HIC_I_1 NA
## -------
## regions: 158 ranges and 0 metadata columns
## seqinfo: 15 sequences from an unspecified genome; no seqlengths
HiContacts package
further provides analytical and visualization tools to investigate Hi-C matrices imported as HiCExperiment in R.
Among other features, it provides the end-user with generic functions to annotate topological features in a Hi-C contact map and export them, notably compartments, domains of constrained interactions (so-called TADs) and focal chromatin loops.
HiCool package integrates an end-to-end processing workflow, to generate multi-resolution balanced contact matrices from paired-end fastq files of Hi-C experiments.
Under the hood, HiCool leverages hicstuff and cooler to process fastq files into .mcool files. hicstuff takes care of the heavy-lifting, and accurately filters non-informative read pairs out, to retain only informative contacts.
Two important features of HiCool are:
- Its operability within the
Recosystem. It relies onbasiliskto set up acondaenvironment with pinned versions of each software it needs to align, filter and process read pairs into contact matrices. - Its transparency.
HiCoolgenerates QC checks and logs, all embedded in HTML files to easily inspect the quality of each sample.
fourDNData (read "4DN Data") provides a gateway to
the 4DN data portal.
HiContactsData package
provides toy datasets to illustrate how the HiCExperiment ecosystem works.
We use devtools and testthat for the development workflow. A Makefile is provided for automation. New functions should be documented with roxygen2 comments and associated tests should be added inside tests/testthat/.
- To install the package for development, run
make install. - To run tests, run
make test - To know more, run
make help
For development purposes, we provide a DockerHub-hosted docker image
with HiCExperiment and related packages pre-installed and ready-to-go.
A new image is automatically built on every push.
## To fetch the latest docker image from Docker Hub (for development purposes!)
docker pull js2264/hicexperiment:latest
## To start docker image
docker run -it js2264/hicexperiment:latest /usr/local/bin/ROn top of that, for each release, an extra docker image is built and
uploaded to the Github Container Repository.
## To fetch release-specific docker image from Github Container Repo
docker pull ghcr.io/js2264/hicexperiment:0.99.9
## To start docker image
docker run -it ghcr.io/js2264/hicexperiment:0.99.9 /usr/local/bin/R