Home

The Trinity Cancer Transcriptome Analysis Toolkit (CTAT) aims to provide tools for leveraging RNA-Seq to gain insights into the biology of cancer transcriptomes. Bioinformatics tool support is provided for mutation detection, fusion transcript identification, de novo transcript assembly of cancer-specific transcripts, lincRNA classification, and foreign transcript detection (viruses, microbes). CTAT is funded by the National Cancer Institute Informatics Technology for Cancer Research (NCI ITCR) program. Software tools and pipelines developed as components of Trinity CTAT are described below with links to the corresponding open source software, documentation, and tutorials.
CTAT-Mutations Pipeline is a variant calling pipeline focussed on detecting mutations from RNA sequencing (RNA-seq) data. It integrates GATK Best Practices along with downstream steps to annotate, filter, and prioritize cancer mutations. This includes leveraging the RADAR and RediPortal databases for identifying likely RNA-editing events, dbSNP for excluding common variants, and COSMIC to highlight known cancer mutations. Finally, CRAVAT is leveraged to annotate and prioritize variants according to likely biological impact and relevance to cancer.
The Trinity CTAT Mutations Pipeline is available at https://github.com/NCIP/ctat-mutations/wiki
We developed DISCASM to assist in the de novo assembly of cancer-specific transcripts. DISCASM restricts de novo transcriptome assembly to those reads that map discordantly or fail to map to the reference genome sequence. Such transcripts are enriched for those that target regions that are restructured or altogether missing from the human reference genome, such as fusion transcripts or those derived from foreign sources (viruses, microbes). Installation of this tool can be through conda or Galaxy toolshed as well.
Detection of cancer fusion transcripts in CTAT is a multi-pronged process involving the use of several alternative individual methods for predicting fusions followed by in silico validation and annotation. Software tools developed as part of CTAT include STAR-Fusion as a highly efficient reference genome read-mapping approach, and DISCASM/GMAP-fusion to leverage de novo transcriptome assembly for fusion detection. In addition, we leverage PRADA and SOAPfuse as additional accurate sources of fusion predictions.

All predicted fusions are subject to in silico validation using our CTAT FusionInspector, which re-evaluates the evidence for fusions predicted by any of the above methods, re-scores the predictions, and uses Trinity to de novo reconstruct likely fusion transcript sequences.
FusionInspector wiki explains conda and Galaxy toolshed installation. GMAP-fusion wiki explains conda and Galaxy toolshed installation. STAR-Fusion wiki explains conda and Galaxy toolshed installation. DISCASM wiki explains conda and Galaxy toolshed installation.
For identification and classification of long noncoding RNAs, we developed Slncky. From a set of reconstructed transcripts, slncky identifies a high-quality set of lncRNA candidates and searches for conserved lncRNAs using a sensitive noncoding aligning method. Trinity genome-free de novo reconstructed transcripts or genome-based transcript reconstructions can be leveraged as input.
lncrna wiki explains conda and Galaxy toolshed installation.
For foreign transcript detection, we leverage Centrifuge and Kraken, leveraging RNA-Seq reads and Trinity-reconstructed transcripts. Our efforts here are being carried out in collaboration with the group of Steven Salzberg at JHU.
ctat-metagenomics wiki explains conda and Galaxy toolshed installation.
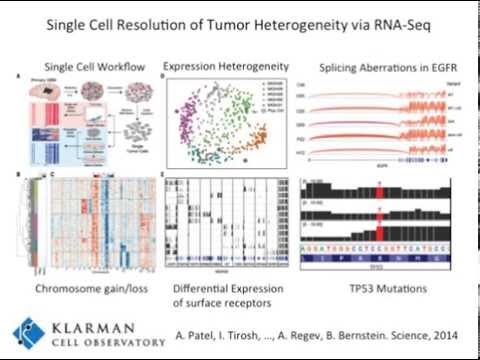
Analysis of single cell transcriptome data to better understand cancer heterogeneity is a growing focus of Trinity CTAT. We are working to update our existing computational components to better leverage single cell transcriptome data, including identifying mutations and fusion transcripts that contribute to tumor heterogeneity.
Among these efforts, we developed an application inferCNV to identify largescale copy number variations (CNV) evident from single cell expression data. Many more contributions are expected to follow shortly.
For Trinity CTAT applications, we aim to enable installation from a variety of sources:
- Software releases from GitHub (minimum for all projects)
- git cloning the 'master' branch from GitHub, which should reflect the most current release.
- Bioconda
- Docker, Singularity
- the FireCloud cloud computing framework
- Galaxy: Galaxy Main or Trinity CTAT Galaxy
See each of the separate project repos for tool-specific installation options.
Contact us via our google group: https://groups.google.com/forum/#!forum/trinity_ctat_users
Trinity CTAT is funded by the National Cancer Institute Informatics Technology for Cancer Research
Our efforts related to building a Trinity Cancer Transcriptome Analysis Toolkit are described in this Youtube screencast: