Home
VirusIntegrationFinder (ctat-VIF) is a module of the Trinity Cancer Transcriptome Analysis Toolkit and used to identify virus insertion sites within the human genome. VIF leverages the STAR aligner to identify chimeric NGS read alignments involving the human genome and a database of viral sequences.
ctat-VIF operates to:
- capture evidence of virus-matching reads (consistent with an infection)
- identify and quantify evidence for viral insertion sites in the human genome
- provide interactive visualizations for evidence of virus-mapped reads and virus insertion sites
See VIF Installation Instructions wiki page for details.
ctat-VIF requires as input Illumina NGS reads, which can be DNA-seq or RNA-seq, and a CTAT genome lib supplemented with viral genomes.
The simplest execution would be:
ctat-vif --left_fq reads_1.fastq \
--right_fq reads_2.fastq \
--sample_id my_sample_name \
-O output_directory \
--genome_lib_dir /path/to/ctat_genome_lib_dir
run 'ctat-vif --help' for full usage info.
STAR alignment step involved here generally requires around 45 GB RAM.
ctat-VIF includes tab-delimited summaries of counts of reads corresponding to the virus insertion sites, counts of reads mapping to the different viral genome targets, and various plots and interactive genome views for interrogating these data. Outputs are detailed below, all based on the included sample data and involve human papillomavirus (HPV).
Output file 'vif.insertion_site_candidates.tsv' contains the following insertion site candidates in tab-delimited format and described below:
chrA coordA orientA chrB coordB orientB prelim.primary_brkpt_type prelim.total split span total
chr11 117344940 + HPV16 1215 + Split 117 33 79 112
chr18 73320805 + HPV16 7409 - Split 168 31 72 103
chr6 64935774 + HPV16 4034 - Split 177 30 70 100
HPV16 4029 + chr3 937922 + Split 1 0 0 0
HPV16 1215 - chr11 108577428 + Split 1 0 0 0
chr2 32018732 + HPV16 1215 + Split 1 0 0 0
chr1 218562612 + HPV16 1215 + Split 1 0 0 0
The coordinates and orientation of the virus fragment are indicated. The 'prelim' columns represent preliminary counts of reads (see algorithmic details below), and the final tally of evidence reads are in the final columns.
The split reads represent those individual reads that align partly to the human genome and partly to the viral genome. Spanning reads are paired-ends where one read maps entirely to the human genome and the other entirely to the viral genome and bridge the insertion site.
The total counts are plotted as 'vif.insertion_site_candidates.genome_plot.png' and as shown below:

Of course, if there are few reads supporting an insertion site, the evidence is very weak and it could be just noise. High read counts are more likely to correspond to valid insertion sites - but examine the reads in the interactive genomics viewer to ensure (shown below).
An interactive igv-reports html file 'vif.html' is included for navigating the read alignment evidence supporting each insertion site. Simply open the html file in your web browser and begin navigating.
At the top of the page you'll have the data table above. Just click on an entry of interest and it will provide the corresponding region in the web-based IGV panel like so:
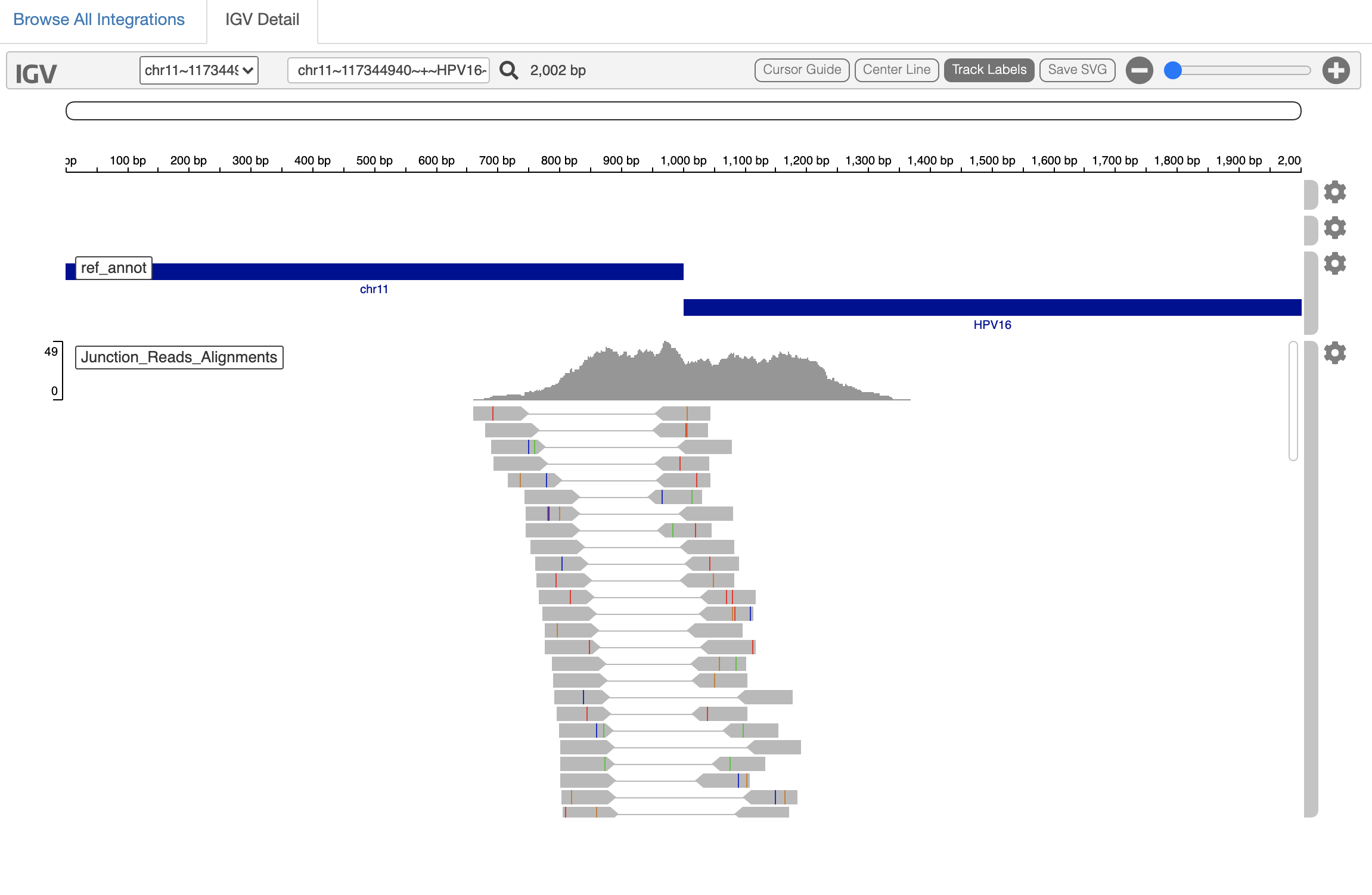
The region of the human chromosome at the putative site of virus insertion is shown along with the reads aligning as split or spanning reads and providing evidence.
beware of low complexity alignments that could be red-herrings... Such regions will be flagged in a future software release.
Even if there isn't strong evidence for virus insertions, we can still detect reads aligning to the target virus sequences and provide interactive visualizations.
File 'vif.virus_read_counts_summary.tsv' contains a tab-delimited summary of read counts corresponding to each viral genome:
virus seqlen mapped chim_reads
HPV16 7906 4546 466
The 'vif.virus.igvjs.html' provides an interactive igv-reports view, tabulating the counts of reads per virus above, and yielding the IGV view for the entry you select, such as below:
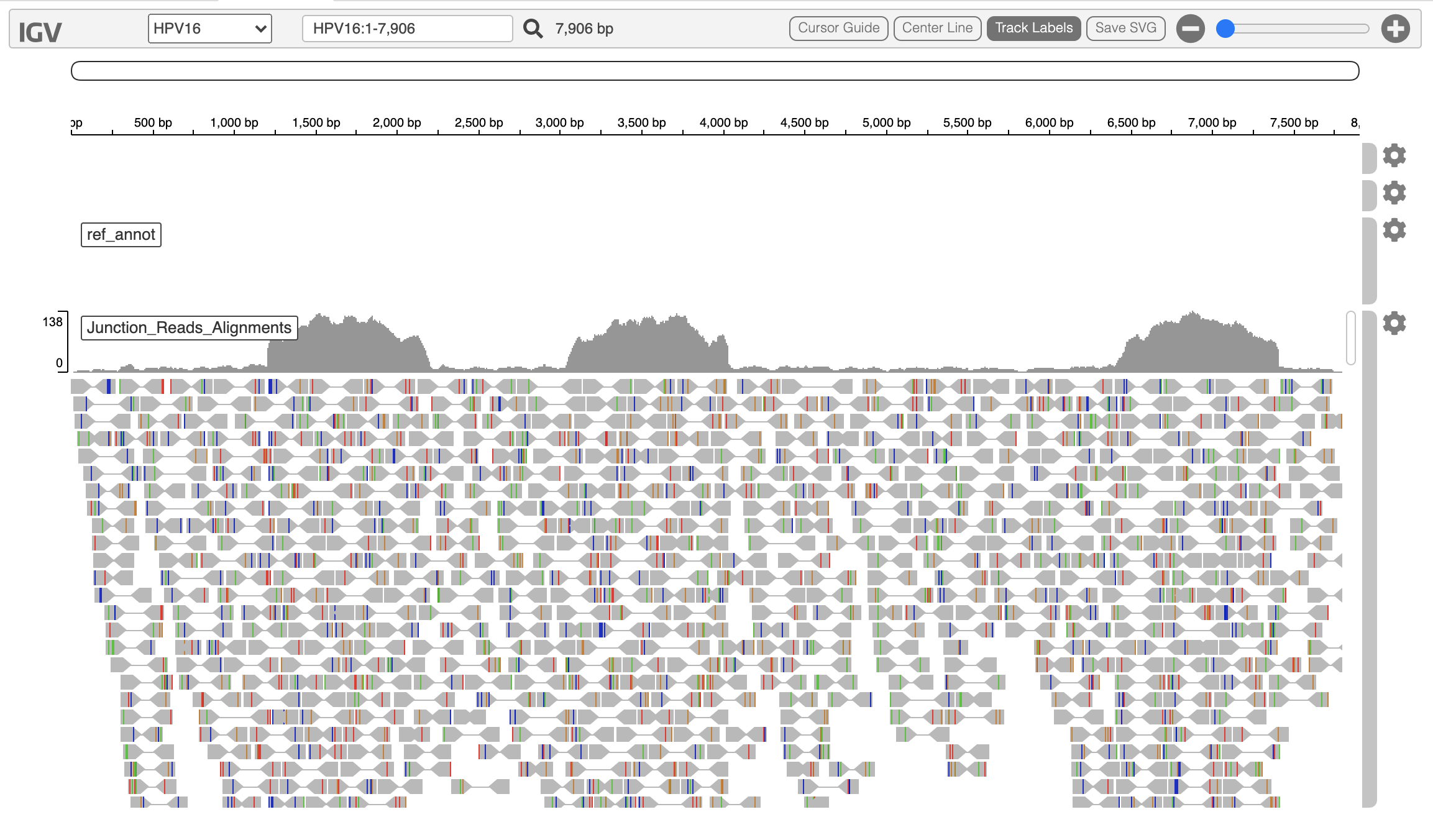
Note, the IGV html is limited to showing about 100x coverage of reads across the genome. This avoids overloading the web browser with an otherwise massive html file. To examine higher coverage and differences in coverage across the genome that exceed this cap, please load the viral-aligned bam file into desktop IGV and interrogate further.
Contact us via our google group: https://groups.google.com/forum/#!forum/trinity_ctat_users
Trinity CTAT is funded by the National Cancer Institute Informatics Technology for Cancer Research
Our efforts related to building a Trinity Cancer Transcriptome Analysis Toolkit are described in this Youtube screencast:
VIF was developed in collaboration with Cristina Montagna, Jack Lenz, and Anne Van Arsdale at Albert Einstein College of Medicine, along with Brian Haas and Joshua Gould at the Broad Institute, and Alexander Dobin at Cold Spring Harbor Laboratory.