Create pseudohaploid assemblies from a partially resolved diploid assembly
Mike Alonge, Srividya Ramakrishnan, and Michael C. Schatz
When assembling highly heterozygous genomes, the total span of the assembly is often nearly twice the expected (haploid) genome size, which is indicative of the assembler partially resolving the heterozygosity. This creates many duplicated genes and other duplicated features that can complicate annotation and comparative genomics. This repository contains code for post-processing an assembly to create a pseudo-haploid representation where pairs of contigs representing the same homologous sequence were filtered to select only one representative contig. The approach is similar to the approach used by FALCON-unzip for PacBio reads or SuperNova for 10X Genomics Linked Reads. As with those algorithms, our algorithm will not necessarily maintain the same phase throughout the assembly, and can arbitrarily alternate between homologous chromosomes at the ends of contigs. Unlike those methods, our method can be run as a stand-alone tool with any assembler.

Oveview of Pseudo-haploid Genome Assembly (a) The original sample has two homologous chromosomes labeled orange and blue. (b) In the de novo assembly, homologous regions containing higher rates of heterozygosity are split into distinct sequences (orange and blue), while regions with low rates or no heterozygous bases are collapsed to a single representative sequence (black). (c) Our algorithm attempts to filter out redundant contigs from the other homologous chromosome, although the phasing of the differ contigs may be inconsistent. Figure derived from [8]
Briefly, the algorithm begins by aligning the genome assembly to itself using the whole genome aligner nucmer from the MUMmer suite. We recommend the parameters nucmer -maxmatch -l 100 -c 500 to report all alignments, unique and repetitive, at least 500bp long with a 100bp seed match. We further filtered these alignments to those that are 1000bp or longer using delta-filter (also part of the MUMmer suite). We also recommend the sge_mummer version of MUMmer so the alignments can be computed in parallel in a cluster environment: sge_mummer github although this will produce identical results to the serial version. Finally we recommend filtering the alignments to keep those that are 90% identity or greater, to filter lower identity repetitive alignments while accommodating the expected rate of heterozygosity between homologous chromosomes while accounting for local regions of greater diversity.
Next, the alignments were examined to identify and filter out redundant homologous contigs. We do so by linking the individual alignments into “alignment chains”, consisting of sets of alignments that are co-linear along the pair of contigs. Our method was inspired by older methods for computing synteny between distantly related genomes, although our method focuses on the problem of identifying homologous contig pairs as high identity long alignment chains. As we expect there to be structural variations between the homologous sequences, we allow for gaps in the alignments between the contigs, although true homologous contig pairs should maintain a consistent order and orientation to the alignments. Specifically, in the alignments from contig A to contig B, each aligned region of A forms a node in an alignment graph, and edges are added between nodes if they are compatible alignments, meaning they are on the same strand, and the implied gap distance on both contig A and contig B was less than 25kbp but not negative. Our algorithm then uses a depth first search starting at every node in the alignment graph to find the highest scoring chain of alignments, where the score is determined by the number of bases that are aligned in the chain. Notably, if a repetitive alignment is flanked by unique or repetitive alignments, such as the orange sequence in Contig B below, this approach will prefer to link alignments that are co-linear on Contig A. We find this produces better results than the filtering that MUMmer’s delta-filter can perform, which does not consider the context of the alignments when identifying a candidate set of non-redundant set of alignments.

Alignment Chain Construction (a) Pairwise alignments between all contigs are computed with nucmer. Here we show just the alignments between contigs A and B. (b) An alignment graph is computed where each aligned region of A forms a node, with edges between nodes that are compatible on the same strand, in the same order, and no more than 25kbp between them. (c) The final alignment chain is selected from the alignment graph as the maximal weight path in the alignment graph.
With the alignment chains identified between pairs of contigs, the last phase of the algorithm is to remove any contigs that are redundant with other contigs originating on the homologous chromosome. Specifically, it evaluates the contigs in order from smallest to longest, and computes the fraction of the bases of each contig that are spanned by alignment chains to other non-redundant contigs. If more than X% of the contig is spanned, it is marked as redundant. This can occur in simple cases where shorter contigs are spanned by individual longer contigs as well as more complex cases where a contig is spanned by multiple shorter non-redundant contigs. We recommmend you evaluate several cutoffs for the threshold of percent of the bases spanned.
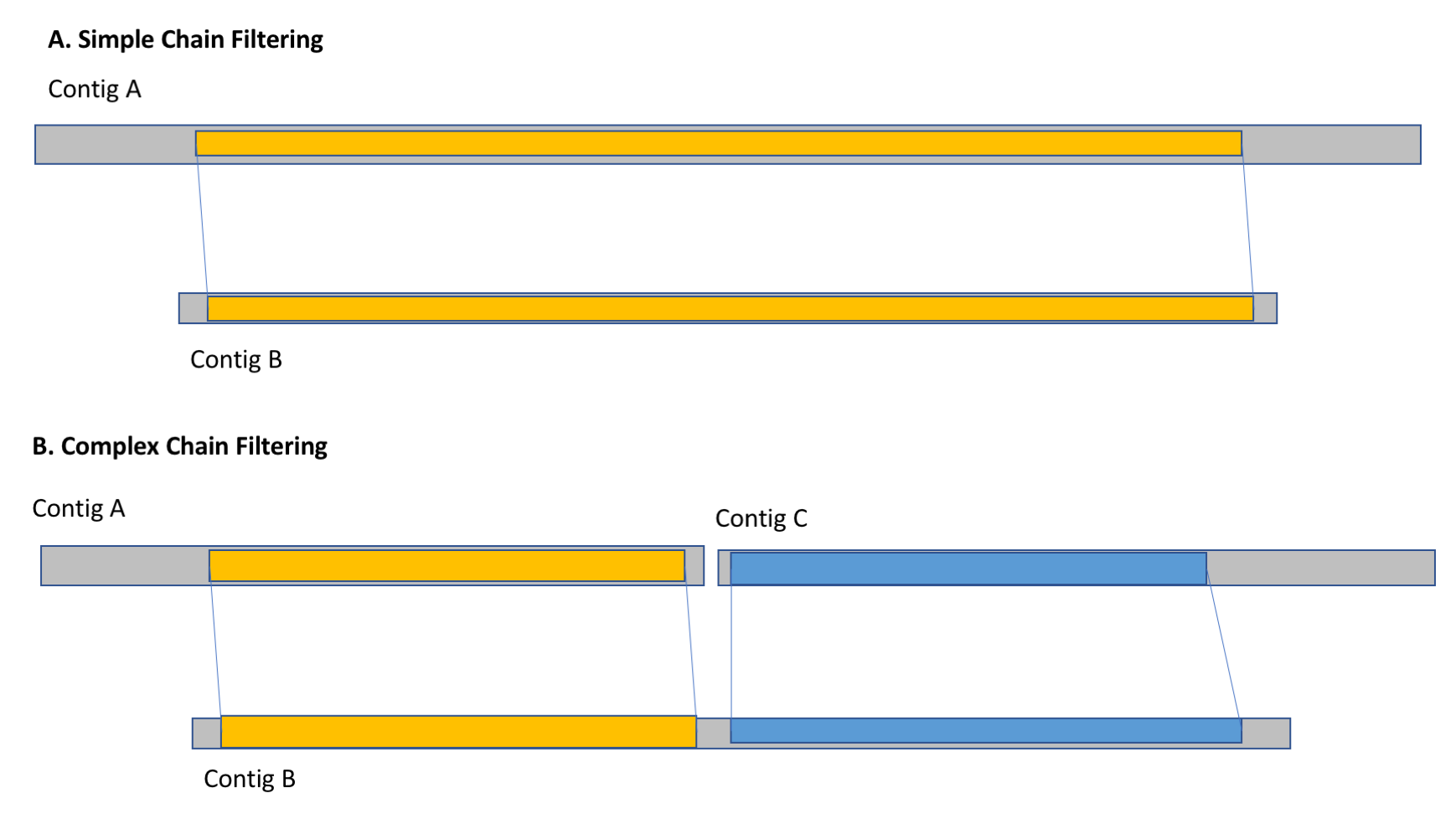
Chain Filtering (a) In simple cases, short contigs (contig A) are filtering out by their alignment chains to longer non-redundant contigs (contig B). (b) In complex cases, a contig (contig B) is filtered out because the total span of the alignment chains to multiple non-redundant contigs (contigs A and C) span more than X% of the bases.
Make sure MUMmer is installed and the binaries are in your path. We recommend version 3.23 although others may work.
Then download the pseudohaploid code:
$ git clone https://github.com/schatzlab/pseudohaploid.git
There is nothing else to install.
The main script to run is create_pseudohaploid.sh. This is a simple bash script to simplify the steps of aligning the genome to itself, filtering the alignments, constructing and analyzing the alignment chains, and then creating the final pseudohaploid assembly. The usage is:
$ create_pseudohaploid.sh assembly.fa outprefix
The test directory has a smalll script to run this comman on a small simple example. If everything is working well you should see:
$ cd test
$ ./run_tests.sh
Running the simple example
Generating pseudohaploid genome sequence
----------------------------------------
GENOME: simple.fa
OUTPREFIX: ph.simple
MIN_IDENTITY: 90
MIN_LENGTH: 1000
MIN_CONTAIN: 93
MAX_CHAIN_GAP: 20000
1. Aligning simple.fa to itself with nucmer
Original assembly has 2 contigs
2. Filter for alignments longer than 1000 bp and below 90 identity
3. Generating coords file
4. Identifying alignment chains: min_id: 90 min_contain: 93 max_gap: 20000
Processing coords file (ph.simple.filter.coords)...
Processed 6 alignment records [4 valid]
Finding chains for 2 contigs...
Found 2 total edges [0.000 constructtime, 0.000 searchtime, 4 stackadd]
Looking for contained contigs...
Found 1 joint contained contigs
Printed 1 total contained contigs
5. Generating a list of redundant contig ids using min_contain: 93
Identified 1 redundant contig to remove in ph.simple.contained.ids
6. Creating final pseudohaploid assembly in ph.simple.pseudohap.fa
Pseudohaploid assembly has 1 contigs
Note the create_pseudohaploid.sh script is just a simple bash script so can be easily editing or incorporated into a larger pipeline. You can also swap out steps, such as replacing nucmer with sge_mummer to use a grid to compute the self alignments.
To demonstrate the capabilities of our new Pseudohaploid method, we applied these techniques to a highly heterozygous sample of Arabidopsis thaliana, an F1 hybrid of Col-0 and Cvi-0 that was previously sequenced as part of the FALCON-unzip paper. For this analysis, we downloaded 116x coverage of PacBio reads (read N50 length=17,474) of the F1 genome from the SRA under accession SRX1715706. We then assembled the reads using Canu using parameters optimized for heterozygous samples. The total size of the raw Canu assembly was substantially larger than the expected haploid genome size: the total assembly size was 214.7Mbp, whereas the haploid genome size is ~135Mbp according to the latest estimates from The Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/portals/genAnnotation/gene_structural_annotation/agicomplete.jsp).
We then applied the Pseudohaploid method to the assembly. This reduced the total size of the assembly from 214.7Mbp to 143.5Mbp, and increased the contig N50 size from 350kbp to 950kbp by reducing the number of contigs from 2074 to 505. Then using the high quality TAIR10 reference genome, we investigated the quality of both the raw and Pseudohaploid assemblies. Using BUSCO, we found the reference genome contained 1356 complete BUSCOs genes, of which 1348 were single-copy, and 8 were duplicated. We found the raw Canu assembly contained a large fraction of duplicated genes, and overall it contained 1355 complete BUSCOs, although only 711 were single-copy, and 644 were duplicated. In contrast the Pseudohaploid assembly substantially reduced the number of duplicate genes, and contained a total of 1355 complete BUSCOs, of which 1240 were single-copy, and only 115 duplicated (an 83% reduction).
Furthermore, by aligning the raw Canu and Pseudohaploid assemblies to the reference TAIR10 assemblies using nucmer using the parameters “-maxmatch -l 100 -c 500”, we found that 1.6Mbp (1.4%) of the TAIR10 assembly was not represented in the Canu assembly, and 4.2Mbp (3.5%) was not represented in the Pseudohaploid assembly as computed by the MUMmer tool dnadiff in the “AlignedBases” field. We also found that 19.0 Mbp of the raw Canu assembly and 14.1Mbp of the Pseudohaploid assembly were unaligned to the reference genome. However, the reference TAIR10 assembly was assembled from the Col-0 accession, and the portions that do not align are chiefly due to the pseudo-haploid representation that will alternate between the Col-0 and Cvi-0 haplotypes. To assess this, we also aligned a high quality (N50 size=7.9Mbp) Cvi inbred assembly created with the FALCON assembler to the TAIR10 reference using nucmer using the same parameters as above. From this, we find that 17.3Mbp (14.5%) of the reference is also not found in the Cvi assembly and the Cvi assembly contains 17.7Mbp not found in the reference highlighting the widespread structural variations between the accessions. We also found that the vast majority (94.5%) of the bases from the Pseudohaploid assembly that were not aligned to the reference genome could be successfully aligned to the Cvi assembly using the same parameters.
Overall, the Pseudohaploid method was highly effective: it removed 71Mbp of redundant sequences from the raw Canu output to substantially improve the fraction of unique genes while only marginally decreasing the sequences from the reference present in the pseudohaploid assembly. The datafiles for these assemblies are available here: http://labshare.cshl.edu/shares/schatzlab/www-data/pseudohaploid/arabidopsis/