DRAFT: BAQ avoidance #1363
DRAFT: BAQ avoidance #1363
Conversation
|
The full depth SynDip chr1 (approx 45x). Time to mpileup|call was 49m28s, vs 105m16s for the standard mpileup method (although I can't recall whether that benchmark was before my other mpileup speed gains).
This one is also interesting as it shows with sufficient depth you can gain higher TP rates (at the cost of modest FP) by not using BAQ at all. You need to be filtering for QUAL>=60ish or higher before using BAQ is actually beneficial (for SNPs). |

df273e4 to
100908d
Compare
de57c12 to
a580a04
Compare
|
@pd3 There's lots of experimentation gone on here, and it needs more evaluation still. However I think it's in a state suitable for you to explore at least. By default BAQ is enabled as before. In addition to the |
|
An example from GIAB HG002 evaluation, including GATK 4 for fun. (All tools with some complex filtering rules applied.) The blue bars by the key are wall-clock time. CPU time is marginally more for bcftools (mpileup|call), and about 80% more for GATK due to threading.
|

|
Latest batch of changes, focusing on Indel calling accuracy, but also demonstrating the effect of changing several default parameters. I've left IMF at 0.05, but my "filtered" values in the tables below has it at 0.1. I think it's maybe worth changing that default too, but it's worthy of debate as it's something we can trivially post-filter on. This experiment is 60x HG002 GIAB sample. Tests are still running, so this is just ~150Mb worth of chr1. When it's finished I'll produce some plots. Here FP = false positives, FN = false negatives and TP = true positives, with true/false defined as matching position and sequence. GT is the genotype errors, so the call was correct but labelled 0/1 when it should have been 1/1 (or vice versa). An incorrect GT but otherwise correct is still included in the TP column, so a more strict definition could be to subtract GT from TP and add it to FN/FP (or maybe half add/subtract if we deem a het vs hom call to be 1/2 correct, 1/2 incorrect). 1.11 defaults (-h100 -m1 -Q13) Not too bad on indels, but it's made a pretty poor job of indels, both in FN and FP. 1.11 -Q1 Dropping Q13 to Q1 is slightly beneficial. (Q0 isn't as it nullifies BAQ.) 1.11 -h500 -m2 -Q1 (Note -m2 is less strict than IDV<2; see unfilt col) Adding -h500 has a huge reduction to FN rates. GT errors rise though. Our filtering is IDV<3 so -m2 is irrelevant, but the unfiltered first column shows the remarkable drop in FP from ~17% to 3%. The default of -m1 is just excessive. This branch Note using Q70 instead of Q50 filtering here to get ballpark same TP/FP SNP range. Biggest change is the hew code has rescued those GT errors, dropping from 19% down to just 1%. Indel FN has also more or less halved, and even unfiltered data is considerably more accurate. The only downside is a slight increase in FP post-filtering (pre-filter it's better). SNPs have marginally increased across the board vs 1.11 (including the modified options). That's possibly an impact of better indel calling, but also maybe -D vs full-BAQ mode rescuing some missing calls. This branch with improved filtering rules Finally with considerably more complex filtering rules we see a better tradeoff of FP vs FN is possible with indels, where the QUAL field is a poor proxy for accuracy (these filtering rules use it sparingly). |
BAQ is the dominant CPU time for mpileup. However it's not needed for the bulk of reads. We delay BAQ until we observe that a read has an overlap with others containing an indel. No indels means no BAQ. Furthermore, if we do have an indel, but this particular read spans the indel with more than 50bp either side then also don't realign it. This is based on the observation that most misalignments occur near the ends of reads where the optimal pair-wise alignment (read vs ref) differs from the optimal multiple alignment (read vs all others). Evaluations so far show this is around a 1.5 to 2-fold speed increase (depending on tuning parameters), while simultaneously slightly increasing SNP calling accuracy. The latter appears to be due to BAQ in non-indel regions sometimes reducing all reads to QUAL 0 in low complexity areas, blocking SNP calling from working. SNPs flanking indels also get blocked for similar reasons.
See ~jkb/work/samtools_master/htslib/_BAQ for results =S as-is and =T is to #if 1 the REALN_DIST part. At 45x They're both slightly behind =L (previous commit) for high QUAL>= filters (favouring more FN over fewer FP), but ahead on lower QUAL thresholds, eg >=50. T is like S but more so, with vastly reduced FN rate. It's somewhere between with-BAQ and without-BAQ in that regard. T is also best for indels. At 15x both S and T are behind L (L > S > T) for all QUAL thresholds on SNPs, but once again on indels T is best. TODO: Consider adding a depth filter so low depth portions are more akin to =L and higher depth to =S (or T)?
Both now have a depth check as BAQ is more vital on shallow data. Also for =T added a limit check to soft-clip frequency. If we have a lot we still use BAQ even for reads spanning the indel.
The previous calc_mwu_bias returns values in the range 0 to 1.
In experimentation I can find little correlation between these values
and the likelihood of a variant being true or false. This is odd,
given GATK's MQRankSum does a good job here.
It appears it's the squashing of Mann Whitney U score to this range
which may be penalising it, but perhaps there is something different
in implementations too? (I started again from scratch based on
Wikipedia.)
This new function takes the same approach as GATK, implementing the
Z-score version where a result of 0 means both distributions are
comparable and above / below zero is used to indicate how extreme the
U score is in either direction. Under testing appears to offer better
discrimination between true and false SNPs, particularly for MQB. For
clarity, we have new MQBZ, RPBZ, BQBZ and MQSBZ fields as the range of
values differs so old filtering rules would be inappropriate.
On a test set, this filter rule seemed to work effectively:
-e "QUAL < $qual || DP>$DP || MQB < -4.5 || RPB > 4 || RPB < -4"
On Chr22 of HG002.GRCh38.60x.bam this gave for 3 different values of
$qual:
SNP Q>0 / Q>=0 / Filtered
SNP TP 39777 / 39777 / 39769
SNP FP 542 / 542 / 483
SNP FN 1316 / 1316 / 1324
InDel TP 4998 / 4998 / 4981
InDel FP 983 / 983 / 52
InDel FN 1945 / 1945 / 1962
SNP Q>0 / Q>=100 / Filtered
SNP TP 39777 / 39567 / 39559
SNP FP 542 / 94 / 71
SNP FN 1316 / 1526 / 1534
InDel TP 4998 / 3934 / 4297
InDel FP 983 / 756 / 22
InDel FN 1945 / 3009 / 2646
SNP Q>0 / Q>=200 / Filtered
SNP TP 39777 / 38074 / 38068
SNP FP 542 / 36 / 27
SNP FN 1316 / 3019 / 3025
InDel TP 4998 / 3200 / 2643
InDel FP 983 / 570 / 10
InDel FN 1945 / 3743 / 4300
Filtered is as per Q>=x but with the additional MQB / RPB filters.
Versus naive QUAL filtering, at Q>=100 it removes 24.5% of the FP
while increasing FN by 0.5%.
Indel filtering is essential to remove the excessive FPs and used:
-e "IDV < 3 || IMF < 0.08 || DP>$DP || QUAL < $qual/5 || \
((5+$qual/6)/(1.01-IMF) < $qual && IDV<(3+$DP*$qual/1000))"
[Something like this needs folding into the indel QUAL score as by
itself it is a very poor indicator of quality. It's still not up to
par even with it, but that's for a future project.]
NB: Above scores are also with a modified BAQ calculation, to come in
another PR.
The old p(U) calc gave eg <-4 and >4 both as near 0, but for some calculations we expect a bias in one direction and not the other. Eg MQB (mapping quality bias) is skewed as REF matching reads have higher MQUAL and ALT matching reads. The U and Z scores are now selectable via #define using the same function, but U is default for compatibility.
This looks for REF / ALT bias in the numbers of soft-clipped bases near variants.
By default it is as before, and -B still disables completely. If we like how it works though it's possible we could make this the default and add a +B for all-BAQ, -B for no-BAQ and have the default inbetween as auto-select BAQ.
The fixed indel size code is (currently) commented out. It's quite slow for now (needs caching), but more importantly it didn't seem to help hugely, especially on deep data. Updated the REALN_DIST code a bit. It's now 40 instead of 50, which helps (especially on deep data) and compensated by tightening the minimum has_clip fraction for lower depth data as BAQ is more important in shallow regions.
Or at least doesn't help universally well. This simplifies things, while being generally a reasonable set of heuristics.
This is a multiplicative factor (albeit 1/x) on the BAQ score in the indel caller. Given the constant, arbitrary, cap of 111 this is more or less equivalent to permitting higher scores. The effect is to be more permissive in calling, giving greater recall rate at a cost of slightly reduced precision. A value of 1 would match the previous computation, but the new default is 2 (=> score * 1/2). On a 30x GIAB HG002 chr1 sample filtering a Q20 this removes 1/3rd of the false negatives, but at a cost of about 1/3rd more FP. At 60x it's about -40% FN + 20% FP. (Given the quantity of true calls hugely outweights false, this is a reasonable trade.) Also adjusted the default -h parameter from 100 to 500. This gives a huge reduction in false negative rates (as much as 3 fold), but also usually has a slightly beneficial decrease in FP. This parameter is used to indicate the likely sequencing error rates in homopolymers, which (for Illumina instruments at least) have greatly reduced since the original bcftools indel caller was written. High -h parameters though does marginally harm SNP calling precision, but not massively.
These values weren't captured in the indel code so we can't post-filter on them. It's hard to know what is ALT and what is REF, but for now we take the naive approach of a read that doesn't have an indel is REF and ALT otherwise, irrespective of quality of match. Of these, MQBZ shows some discrimination (< -5 for 30x and < -9 for 150x), but RPBZ and SCBZ have overlapping distributions (although RPBZ may just about have a slim margin worthy of filtering at the right end). That said for both the dist of the true variants extends much further negative than the dist for the false ones, so while we cannot remove FP without removing TP, we could at least use this for computing a refined QUAL value. [BQBZ undergoing testing.]
This is the minimum base quality in the region post BAQ as this was found to be the most robust mechanism.
These are now computed using their own arrays, as there was some leakage with the SNP stats. Also noticed while assessing the new metrics that there is a very high rate of incorrect indel genotypes, particularly HOM to HET. After experimentation, I realised that seqQ governs which candidate types end up in the genotype, and not indelQ. seqQ is now capped at indelQ and no higher. This has had a huge reduction in GT miscalls. [With amended compare_vcf.sh script to now validate GT also matches, in their own output line] 15x GIAB HG002 before: SNP Q>0 / Q>=10 / Filtered SNP TP 257554 / 255433 / 255224 SNP FP 1846 / 1049 / 998 SNP GT 883 / 853 / 882 SNP FN 6756 / 8877 / 9086 InDel TP 42021 / 41651 / 40762 InDel FP 709 / 594 / 347 InDel GT 7326 / 7306 / 7242 InDel FN 3583 / 3953 / 4842 15x GIAB HG002 after: SNP Q>0 / Q>=10 / Filtered SNP TP 257554 / 255433 / 255224 SNP FP 1846 / 1049 / 998 SNP GT 883 / 853 / 882 SNP FN 6756 / 8877 / 9086 InDel TP 43008 / 42651 / 41723 InDel FP 820 / 706 / 454 InDel GT 1098 / 1087 / 1036 InDel FN 2596 / 2953 / 3881 Note reduction to indel FN too (and modest increase in FP). It's even more pronounced on deep data. 150x GIAB HG002 before: SNP Q>0 / Q>=10 / Filtered SNP TP 262711 / 262628 / 262523 SNP FP 1947 / 1621 / 1323 SNP GT 253 / 244 / 239 SNP FN 1599 / 1682 / 1787 InDel TP 44451 / 44435 / 44384 InDel FP 4617 / 4455 / 1279 InDel GT 6333 / 6327 / 6321 InDel FN 1153 / 1169 / 1220 150x GIAB HG002 after: SNP Q>0 / Q>=10 / Filtered SNP TP 262711 / 262628 / 262523 SNP FP 1947 / 1621 / 1323 SNP GT 253 / 244 / 239 SNP FN 1599 / 1682 / 1787 InDel TP 45061 / 45053 / 44992 InDel FP 4798 / 4628 / 1408 InDel GT 448 / 446 / 441 InDel FN 543 / 551 / 612 Here we're seeing a 14 fold reduction in the rate of incorrect indel genotype assignment, and a 2 fold reduction in false negative rates.
Just switching seqQ and baseQ in mpileup.c is easier than the bit twiddling in bam2bcf_indel.c. Also adjusted the default indel-bias down again from 2 to 1. This tweak was needed before, but the change in scoring function makes it better again with the default params.
Also tweak indelQ to still use seqQ. It doesn't help calling (infact it's very marginally poorer), but it grants a use of -h and indel open/extend costs too which had been elided in previous commits.
The Short Tandem Repeat from Crumble has been added to the indel
caller. This is used to calculate the STRBZ INFO metric (not so
useful I think, so maybe to remove) and to skew the indel score
slightly for reads overlapping an indel site that start or end in an
STR.
At low filtering levels I get these results or HG002 chr1:
15x previous:
Q>0 / Q>=10 / Filtered
InDel TP 42256 / 41744 / 41082
InDel FP 601 / 540 / 375
InDel GT 1265 / 1234 / 1191
InDel FN 3348 / 3860 / 4522
15x this:
Q>0 / Q>=10 / Filtered
InDel TP 42354 / 41806 / 41186
InDel FP 554 / 501 / 343
InDel GT 1180 / 1151 / 1121
InDel FN 3250 / 3798 / 4418
That's a decrease to all of FP, GT and FN.
At 150x previous:
Q>0 / Q>=10 / Filtered
InDel TP 44854 / 44838 / 44775
InDel FP 3541 / 3387 / 869
InDel GT 517 / 516 / 507
InDel FN 750 / 766 / 829
At 150x this:
Q>0 / Q>=10 / Filtered
InDel TP 44917 / 44897 / 44890
InDel FP 778 / 753 / 750
InDel GT 412 / 409 / 410
InDel FN 687 / 707 / 714
This is also a decrease to all of FP, GT and FN. In particular
unfiltered FP has a huge drop, so naive usage of bcftools will be
considerably more accurate.
TODO: this clearly works well for indels. Maybe consider adding it
for SNPs too?
This is length / rep_len. The theory being the +score is proportional to the copy number rather than total length, as that is a better indication of the number of different alignment possibilities. In practice it seems slightly better discriminating, although the jury is out whether that's simply due to score being lower on average.
I noticed a few issues while doing this, but they'll be resolved in subsequent commits as it'd be nigh on impossible to identify the changes if included here. This should give identical results to the previous commit, aiding validation that the refactoring hasn't changed anything.
The checking for tbeg/tend was incorrect as one is in ref coords and the other is local to the buffer, making the test basically always true. This just accepts that and removes the condition, as the end result appears to show the code works.
- Remove STRBZ and BQBZ as unuseful stats. - Only compute stats once per indel rather than per type. - Replace some pointless mallocs with fixed arrays as the maximum number of types was already hard-coded. - General code tidyups
These needs new parameters for BAQ (also analous ones in htslib realn.c) as the error models differ with most errors in indels instead of substitutions. Detection of platform is very crude currently - just length. TODO: use @rg PL field and DS containing "CCS". Protect against attempts to call BAQ on negatived sized regions. "tend" can become less than "tbeg" as tend is left-justified and tbeg right-justified when it comes to deletions. If the entire left..right portion is contained within a single cigar D op, then tend<tbeg. Protect against excessive BAQ band widths, which is both very costly in CPU time (with ONT I saw one single BAQ call take 30 mins), and uses vast amounts of memory. For now this is a hard cutoff, but we may consider making it adjustable. BAQ has minimal benefit to PacBio CCS and it's untested on ONT as it triggered the out-of-memory killer. Also add a maximum read length for BAQ (--max-read-len) so we can trivially simply disable it for all long-read data while keeping it enabled on short reads, eg mixed data sets with ONT, PacBio and Illumina.
The enlarged INDEL_WINDOW_SIZE, beneficial in Illumina data, is a CPU hog for PacBio as we get many more candidate indels (unless we bump the -m parameter). However it also turns out to be slightly better to have a lower window size too on CCS.
It only needs to check first few and last few elements to obtain soft-clip data, as it's trying to turn absolute position / length into clip-adjusted position / length. This function can easily be 30%+ of the CPU time when using long read technologies.
Commit 0345459 rewrote this so it worked well on PacBio CCS data, but in the process it changed the algorithm slightly for SR data. The "ncig > 1" check is back and we now also don't do ignore hard/soft-clips on short read data too. These are necessary to avoid false positives, which jumped with illumina data after the change before.
"INDEL" tag is added on call->ori_ref < 0, but the check was wrongly just non-zero. This harmed the generation of SNP filter tags.
This was already done for long reads, but it turns out the second call is detrimental for the short read data sets I tested too.
|
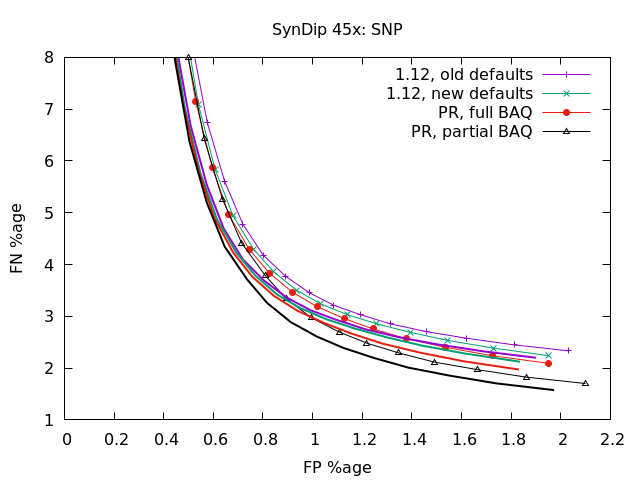
Latest round of tests, after upgrading to cope with PacBio CCS data (plots to follow). This is SynDip at 15x and 45x coverage. The thin lines with symbols, as shown in the keys, are false negative vs false positive using a very strict definition of correctness. The variant must be the same coordinate, the same sequence, and the same genotype (ie 0/0, 1/1, 0/1, etc). If the position and sequence is correct but the genotype incorrect, it adds half to FN and half to FP (as generally it's got one allele correct and one wrong). The solid lines without symbols, but using the same colours, are without that penalty of requiring the correct genotype too. It's noticably different on indels for the 1.12 as it had a high GT failure mode.
At 15x, SNPs don't show much difference between this PR and release 1.12 with updated default parameters. With INDELs however there is a very significant shift once genotypes are checked, and even without that there is a shift towards fewer FN with the new code. Note full vs partial BAQ has no impact on the indel caller.
At 45x, it's much the same as 15x only more exaggerated so it's easier to see the impact. In particular partial BAQ with this PR is a significant reduction in FN rate for the SNP caller at the lower quality values (focusing on recall over precision). |



57fe763 to
98bbd48
Compare
|
This data set is GIAB HG002, subsampled to 15, 60 and 150x (from 300x).
At 15x there's very little in it with any tool / parameters for SNPs. There is some marginal gain for the new code, but only visible if you zoom in a lot. INDELs show the same gain in GT assignment accuracy we saw before, and once again is a little more sensitive even if we ignore the GT issue.
At 60x it's clear that partial BAQ is better than full BAQ for all SNPs except perhaps at very stringent filtering levels, but both with/without are considerably more sensitive than the old code even with new parameters. This deeper data really shows the impact of the new default parameters. This is primarily -h500 as the default (-h100) misses around 30% of indels - presumably thinking they're polymerase stutter instead.
150x shows the same characteristics as 60x, only more so. The reduction in false negatives for indels is particularly noticable. |






|
Edit: I'll reanalyse the PacBIO CCS data set again now 1.12 has been released to do a proper side by side comparison once more, and post figures once I have them. Meanwhile, the biggest impact in this PR is changing the defaults to be more appropriate for modern data and the bug fix to improve the indel genotype accuracy. That was a relatively small change (using seqQ instead of indelQ, or vice versa - I forget which way it went). Finally, I recorded speeds too as this is what my original intention was. Improving accuracy was just a happy fluke of analysing that my speedups weren't detrimental, and then researching the large source of missing indels. GIAB HG002
CHM1/CHM13
|
|
Now for some PacBio CCS results. This data set was HG002 from GIAB again, and around 31x depth. I've had to plot these in log scale (Y) because the range was so vast. In particular bcftools 1.12 defaults really don't like CCS at all.
This is messy and hard to see due to the disparity in Y range and also there are multiple lines scrunched on top of one another. Firstly, there are two clear parts - 1.12 with full BAQ (default) vs everything else. 1.12 defaults are just terrible for this data. Fortunately 1.12 with -B to disable BAQ brings it into the mix with this PR. It has slightly higher false positive rate, but the new defaults address that and it's then identical to this PR with defaults or -D (partial BAQ). Why? Because the new PR with full-baq or partial-BAQ is the same as no-BAQ due to a new maximum BAQ-length limit. To force BAQ on, we need to use -M, set here to an arbitrary large value, and then we see a considerable improvement in FP and a slight improvement in FN. As before I also show these figures with GT errors ignored, but the pattern is the same - just shifted.
Indels are a different picture, clustering in 3 locations. Hard to see, but 1.12 with default params, both with and without BAQ, are in the top left corner. The default parameters just don't work for indel calling on 1.12. Next up is 1.12 with new defaults (-h500 -m2 -Q1), which we see rescues a major portion of the indels, but it's still missing > 10%. This PR however is a very significant reduction in both FN and FP. This is because I changed the BAQ parameters as the probabilities for the HMM state change are inappropriate for this technology. They all line up together though as BAQ is forcibly on always for indels. The without-GT-error case has just two lines visible, but it's mirroring the two groups above with data superimoposed, so the bottom left line is all the this-PR data. Speed wise, there were a few surprises:
The 1.12 versions with BAQ were very slow and also huge significant memory up in computing BAQ, because the band size grows considerably making it a large slow matrix. Without BAQ 1.12 is reasonably fast, although note the big difference between "-B" and "-B -m2 ...". The -m2 there is removing a huge portion of candidate indels which get tested for and rejected. This PR speed, with full and partial BAQ (without -M) are the same time, and they'd be much the same as no-BAQ too, due to the length limit being applied meaning no BAQ calcs were actually made. They're slightly behind 1.12 -B, probably due to reserving memory for the potential BAQ qual values. The suprise is I have some more tests running to see what full-BAQ |


There is now a maximum quality value, defaulting to 60 so above Illumina values. This is useful or PacBio CCS which produces abnormally high qualities leading to over-certainty in calls, especially for incorrect genotype assignment. SNP base quality is now the minimum of this base qual plus the base either side. CCS data commonly has a neighbouring low qual value adjacent to in incorrect but high quality substitution. However it turns out this is also beneficial to the HG002 Illumina data. An example of the effect for Illumina 60x data (with adjustment on min QUAL to get the same FN rate as the scores are slightly suppressed now): Before: SNP Q>0 / Q>=20 / Filtered SNP TP 262541 / 262287 / 262283 SNP FP 2313 / 1442 / 1405 SNP GT 287 / 269 / 269 SNP FN 1769 / 2023 / 2027 After: SNP Q>0 / Q>=15 / Filtered SNP TP 262503 / 262298 / 262294 SNP FP 1787 / 1349 / 1312 -6.6% SNP GT 283 / 268 / 268 ~= SNP FN 1807 / 2012 / 2016 -0.5% For 31x PacBio CCS, the quality assignment suppression is more pronounced due to the excessively high QUALs in the bam records, but the FN/FP tradeoff is the same (again tuning to try and compare with ~= FN): Before: SNP Q>0 / Q>=47 / Filtered SNP TP 263847 / 263693 / 263693 SNP FP 4908 / 3089 / 3089 SNP GT 804 / 793 / 793 SNP FN 463 / 617 / 617 After: SNP Q>0 / Q>=15 / Filtered SNP TP 263779 / 263692 / 263692 SNP FP 3493 / 2973 / 2973 -10% SNP GT 509 / 503 / 503 -37% SNP FN 531 / 618 / 618 ~= Finally, added -X,--config STR option to specify sets of parameters tuned at specific platforms. Earlier experiments showed the indel caller on PacBio CCS data needs very different gap-open and gap-extend parameters. It also benefits a bit from raising minimum qual to 5 and because this is a new option we also enable partial realignment by default.
--max-read-len (-M), --config (-X) and --max-BQ.
f7bc9cf to
317e4ca
Compare
As it's held in static memory, it needed to put the default '@' symbol back after nulling it. This of course makes it not thread safe, but we don't have any code to run this in a threaded environment. The alternative is a non-static buffer and memset the appropriate length each time. I haven't tested what overhead that brings.
This disables indels (they're *glacial* to compute at the moment for long-cigar reads and really don't give any real accuracy at present), disables BAQ, and tweaks min(filter) and max(clip) qualities.
The fixes in commit 3c1205c while still here, no longer worked in this branch due to the swapping of seqQ and baseQ. The fix however seemed inappropriate and caused other issues, such as samtools#1446. The new logic is to check p->indel to decide if the read needs b=0 (REF) setting rather than a naive qual check. This greatly reduces the false positive rate of indel calling, especially when deep, however it also increases GT errors a bit. I've tried to get some middle ground where the new seqQ used for REF calls isn't mostly the old qual, but with a partial mix in of the base qual. I don't like heuristics, but it works well on the data sets tested and fixes both the issue addressed by 3c1205c and also samtools#1446, while keeping the FP reduction. Beneficial at 30x, 60x and 150x both filtered and unfiltered (huge win). At 15x it's a small win for unfiltered and a moderate loss for filtered, almost entirely due to higher GT error rate. Possibly this is filtering rules no longer appropriate, or maybe it's simply not good on shallower data. Needs investigation. TODO: the baseQ assignment seems off. We use this for MQBZ, but in testing this has very little filter discrimination power for indels so we don't recommend it. It's possible this is because it's not handling REF/ALT mixes well. It needs investigation to see which of seqQ, baseQ and bam_get_qual()[] values are most appropriate for this stat.
These are all labelled mp5/mp6 to mp6f in the code, with mp6e being chosen. TODO: Cull the others once I'm happier with more testing, but keeping them here for now as documentation of what I've explored. The chosen method generally has around 30% reduction to FP rates over earlier efforts, although this is depth specific.
This modifies some of f4e8f72. It's less useful for Illumina than PacBio-CCS. Infact the old method was detrimental to the SynDip test data. Our strategy now is to check the delta between neighbour and current quality. If neighbour+D is lower than qual then use neighbour+D instead, where D defaults to 30 (15 for PacBio).
(This just removes #if-ed out code.)
|
Closing as replaced by #1474 |
Not every call to BAQ is needed, indeed not every call is beneficial.
This is a work-in-progress DRAFT PR, but I'm making it as a reminder to my post-Christmas self. :-)
Basically it replaces the always-call-
sam_prob_realncode with some analysis on indels and soft-clips in the pileup constructor. Then during the pileup phase, any time we have reads overlapping a certain number of indels / soft-clips we'll trigger the realignment there.The result is we shave off about 1/3rd of the CPU time for mpileup, while simulatenously slightly improving on SNP calling accuracy. See the figure below on a low coverage (15x) copy of SynDip chr1:
Indel calling is marginally harmed, but it's very minimal and it's still better than minus-BAQ.
With BAQ
With partial BAQ
That's all indels and filtered indels (filtering is a must for indels from bcftools).