Integrating Spectra with Python's matchms package

![]()
The SpectriPy package allows integration of Python MS packages into a
Spectra-based MS analysis in R. Python functionality is wrapped into R
functions allowing a seamless integration of the functionality of Python's
matchms package into R. In addition,
functions to convert between R's Spectra objects and Python's matchms
spectrum objects are available to the advanced user or developer enabling to
create custom functions or workflows on Spectra objects in Python and
executing them in R using the reticulate R package.
Instructions to install R and RStudio for the first time are described below, from source https://rstudio-education.github.io/hopr/packages2.html.
To get started with R, you need to acquire your own copy. This appendix will show you how to download R as well as RStudio, a software application that makes R easier to use. You’ll go from downloading R to opening your first R session.
Both R and RStudio are free and easy to download.
R is maintained by an international team of developers who make the language available through the web page of The Comprehensive R Archive Network. The top of the web page provides three links for downloading R. Follow the link that describes your operating system: Windows, Mac, or Linux.
To install R on Windows, click the “Download R for Windows” link. Then click the
“base” link. Next, click the first link at the top of the new page. This link
should say something like “Download R 3.0.3 for Windows,” except the 3.0.3
will be replaced by the most current version of R. The link downloads an
installer program, which installs the most up-to-date version of R for
Windows. Run this program and step through the installation wizard that
appears. The wizard will install R into your program files folders and place a
shortcut in your Start menu. Note that you’ll need to have all of the
appropriate administration privileges to install new software on your machine.
To install R on a Mac, click the “Download R for Mac” link. Next, click on the
R-3.0.3 package link (or the package link for the most current release of
R). An installer will download to guide you through the installation process,
which is very easy. The installer lets you customize your installation, but the
defaults will be suitable for most users. I’ve never found a reason to change
them. If your computer requires a password before installing new progams, you’ll
need it here.
Binaries Versus Source
R can be installed from precompiled binaries or built from source on any operating system. For Windows and Mac machines, installing R from binaries is extremely easy. The binary comes preloaded in its own installer. Although you can build R from source on these platforms, the process is much more complicated and won’t provide much benefit for most users. For Linux systems, the opposite is true. Precompiled binaries can be found for some systems, but it is much more common to build R from source files when installing on Linux. The download pages on CRAN’s website provide information about building R from source for the Windows, Mac, and Linux platforms.
R comes preinstalled on many Linux systems, but you’ll want the newest version of R if yours is out of date. The CRAN website provides files to build R from source on Debian, Redhat, SUSE, and Ubuntu systems under the link “Download R for Linux.” Click the link and then follow the directory trail to the version of Linux you wish to install on. The exact installation procedure will vary depending on the Linux system you use. CRAN guides the process by grouping each set of source files with documentation or README files that explain how to install on your system.
32-bit Versus 64-bit
R comes in both 32-bit and 64-bit versions. Which should you use? In most cases, it won’t matter. Both versions use 32-bit integers, which means they compute numbers to the same numerical precision. The difference occurs in the way each version manages memory. 64-bit R uses 64-bit memory pointers, and 32-bit R uses 32-bit memory pointers. This means 64-bit R has a larger memory space to use (and search through). As a rule of thumb, 32-bit builds of R are faster than 64-bit builds, though not always. On the other hand, 64-bit builds can handle larger files and data sets with fewer memory management problems. In either version, the maximum allowable vector size tops out at around 2 billion elements. If your operating system doesn’t support 64-bit programs, or your RAM is less than 4 GB, 32-bit R is for you. The Windows and Mac installers will automatically install both versions if your system supports 64-bit R.
R isn’t a program that you can open and start using, like Microsoft Word or Internet Explorer. Instead, R is a computer language, like C, C++, or UNIX. You use R by writing commands in the R language and asking your computer to interpret them. In the old days, people ran R code in a UNIX terminal window—as if they were hackers in a movie from the 1980s. Now almost everyone uses R with an application called RStudio, and I recommend that you do, too.
R and UNIX
You can still run R in a UNIX or BASH window by typing the command:
Rwhich opens an R interpreter. You can then do your work and close the interpreter by running q() when you are finished.
RStudio is an application like Microsoft Word—except that instead of helping you write in English, RStudio helps you write in R. I use RStudio throughout the book because it makes using R much easier. Also, the RStudio interface looks the same for Windows, Mac OS, and Linux. That will help me match the book to your personal experience.
You can download RStudio for free. Just click the “Download RStudio” button and follow the simple instructions that follow. Once you’ve installed RStudio, you can open it like any other program on your computer—usually by clicking an icon on your desktop.
The R GUIs
Windows and Mac users usually do not program from a terminal window, so the Windows and Mac downloads for R come with a simple program that opens a terminal-like window for you to run R code in. This is what opens when you click the R icon on your Windows or Mac computer. These programs do a little more than the basic terminal window, but not much. You may hear people refer to them as the Windows or Mac R GUIs.
When you open RStudio, a window appears with three panes in it, as in Figure 1. The largest pane is a console window. This is where you’ll run your R code and see results. The console window is exactly what you’d see if you ran R from a UNIX console or the Windows or Mac GUIs. Everything else you see is unique to RStudio. Hidden in the other panes are a text editor, a graphics window, a debugger, a file manager, and much more. You’ll learn about these panes as they become useful throughout the course of this book. The RStudio IDE for R.
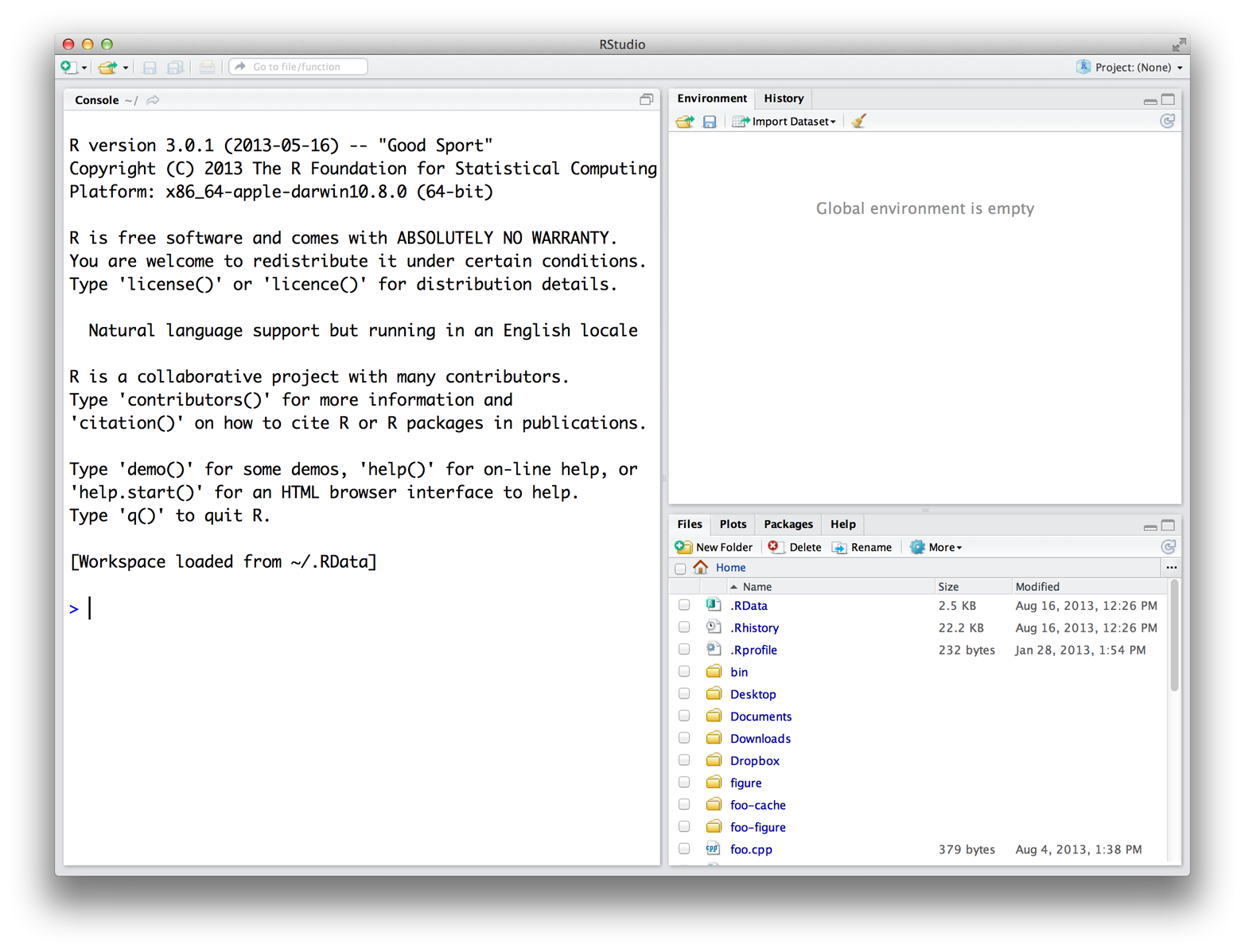
{kind=link}
Figure 1: The RStudio IDE for R.
Do I still need to download R?
Even if you use RStudio, you’ll still need to download R to your computer. RStudio helps you use the version of R that lives on your computer, but it doesn’t come with a version of R on its own.
Now that you have both R and RStudio on your computer, you can begin using R by opening the RStudio program. Open RStudio just as you would any program, by clicking on its icon or by typing “RStudio” at the Windows Run prompt.
Bioconductor is required to install SpectriPy, as described below and from source https://bioconductor.org/install/.
The current release of Bioconductor is version 3.20; it works with R version 4.4.0. Users of older R and Bioconductor must update their installation to take advantage of new features and to access packages that have been added to Bioconductor since the last release.
The development version of Bioconductor is version 3.21; it works with R version 4.5.0. More recent ‘devel’ versions of R (if available) will be supported during the next Bioconductor release cycle.
Once R has been installed, get the latest version of Bioconductor by starting R and entering the following commands.
It may be possible to change the Bioconductor version of an existing installation; see the ‘Changing version’ section of the BiocManager vignette.
Details, including instructions to install additional packages and to update, find, and troubleshoot are provided below. A devel version of Bioconductor is available. There are good reasons for using BiocManager::install() for managing Bioconductor resources.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.20")
SpectriPy uses basilisk to
ensure all required python packages are installed and available (in the correct
version) on each system. basilisk installs a self-contained conda environment,
thus, the SpectriPy package is independent of the system's Python environment.
To install the package use
BiocManager::install("RforMassSpectrometry/SpectriPy")
The status of installation can be easily checked by starting R and entering the following commands.
This command loads the SpectriPy package, if correct installed.
library(SpectriPy)
The concepts and examples can be checked by performing some steps from the package’s vignette, from source https://rformassspectrometry.github.io/SpectriPy/articles/SpectriPy.html.
To install the Spectra
package use following command. This package is needed, as the SpectriPy
package allows integration of Python MS packages into a Spectra-based MS
analysis in R.
BiocManager::install("RforMassSpectrometry/Spectra")
The SpectriPy package provides the compareSpectriPy() function that allows
to perform spectra similarity calculations using the scoring functions from the
matchms Python package. Below all currently supported scoring functions are
listed along with the parameter class that allows selecting and configuring
the algorithm in the compareSpectriPy() function. Additional functions will be
added in future.
- CosineGreedy:
CosineGreedyParam. - CosineHungarian:
CosineHungarianParam. - ModifiedCosineParam:
ModifiedCosineParam.
We next create some simple example spectra and subsequently use the
compareSpectriPy() function to calculate pairwise similarities between these.
library(Spectra)
library(SpectriPy)
## Create a Spectra object with two MS2 spectra for Caffeine.
caf <- DataFrame(
msLevel = c(2L, 2L),
name = "Caffeine",
precursorMz = c(195.0877, 195.0877)
)
caf$intensity <- list(
c(340.0, 416, 2580, 412),
c(388.0, 3270, 85, 54, 10111))
caf$mz <- list(
c(135.0432, 138.0632, 163.0375, 195.0880),
c(110.0710, 138.0655, 138.1057, 138.1742, 195.0864))
caf <- Spectra(caf)
## Create a Spectra object with two MS2 spectra for 1-Methylhistidine
mhd <- DataFrame(
msLevel = c(2L, 2L),
precursorMz = c(170.0924, 170.0924),
id = c("HMDB0000001", "HMDB0000001"),
name = c("1-Methylhistidine", "1-Methylhistidine"))
mhd$mz <- list(
c(109.2, 124.2, 124.5, 170.16, 170.52),
c(83.1, 96.12, 97.14, 109.14, 124.08, 125.1, 170.16))
mhd$intensity <- list(
c(3.407, 47.494, 3.094, 100.0, 13.240),
c(6.685, 4.381, 3.022, 16.708, 100.0, 4.565, 40.643))
mhd <- Spectra(mhd)
We first calculate pairwise similarities between all spectra defined above and
those of caffeine using Spectra's built-in compareSpectra() function.
all <- c(caf, mhd)
res_r <- compareSpectra(all, caf)
res_r
Thus, compareSpectra() returned the pairwise similarity scores (by default
calculated using the normalized dot-product function) between all spectra in
all (rows) and all spectra in caf (columns). compareSpectriPy() works
similar, with the difference that we need to specify and configure the
similarity function (from matchms) using a dedicated parameter object. Below
we calculate the similarity using the CosineGreedy function changing the
tolerance to a value of 0.05 (instead of the default 0.1).
res <- compareSpectriPy(all, caf, param = CosineGreedyParam(tolerance = 0.05))
res
As a result compareSpectriPy returns also a numeric matrix of similarities.
Note also that the first compareSpectriPy call takes usually a little longer
because the Python setup has to be initialized.
Next we use the ModifiedCosine algorithm that considers also differences between the spectra's precursor m/z in the calculation.
res <- compareSpectriPy(all, caf, param = ModifiedCosineParam())
res
Note that for this calculation all spectra precursor m/z values need to be
available, otherwise an error will be thrown. Thus, we should always ensure to
remove spectra without precursor m/z values prior to similarity scoring with
this similarity method. Below we remove the precursor m/z from one of our input
spectra and then show how the Spectra object could be subsetted to valid
spectra for this method.
## Remove precursor m/z from the 3rd spectrum
all$precursorMz[3] <- NA
## Filter the input spectra removing those with missing precursor.
all <- all[!is.na(precursorMz(all))]
compareSpectriPy(all, caf, param = ModifiedCosineParam())
See the package's vignette.
- TODO explain how package is structured
- TODO explain where the code is
- TODO mention coding style
- TODO mention some things that could/should be implemented
Contributions are highly welcome and should follow the contribution guidelines. Also, please check the coding style guidelines in the RforMassSpectrometry vignette.