Usage questions about nglview: Please ask here first [2] #901
Comments
|
Hi! Is there a way to work with the residue indices (0-th based), not the ids, in a selection? Thanks in advance!! |
|
hi @dprada, I don't know yet on top of my head. Let me think a bit. |
|
Hello, thank you for all your work on this great tool. I am trying to calculate and visualize a molecular electrostatic potential, on a specific electron density isosurface, kind of similar to this example with ngl (live example), but using PySCF and nglview, within a Jupyter notebook: from pyscf import gto
from pyscf import dft
from pyscf.geomopt import berny_solver
from pyscf import __config__
from pyscf.tools import cubegen
import nglview as nv
# Default of 3.0 is too small for 6-31G*
setattr(__config__, 'cubegen_box_margin', 4.0)
h2o = gto.M(atom=' O 0.00000000, 0.000000, 0.119748;\
H 0.00000000, 0.761561, -0.478993;\
H 0.00000000, -0.761561, -0.478993',\
basis='6-31G*')
h2o.build()
h2o.verbose = 0
method = dft.RKS(h2o)
method.grids.prune = dft.gen_grid.treutler_prune
method.grids.atom_grid = {"H": (50, 194), "O": (50, 194)}
method.xc = 'b3lypg'
method.scf()
opt_mol = berny_solver.optimize(method)
cubegen.density(opt_mol, 'water_den.cube', method.make_rdm1(), resolution=(1/6))
cubegen.mep(opt_mol, 'water_pot.cube', method.make_rdm1(), resolution=(1/6))
view = nv.NGLWidget()
view.add_component('water_den.cube', color="red")
view.component_0.add_surface(isolevelType="value", isolevel=0.002)
view.add_component('water_pot.cube')
view.component_1.add_surface(color='red')
viewI think I am close to getting this to work, but I am not sure how to use data from the two components together. |
|
Dont' worry @hainm, it is something that can be done combining the chain and residues ids. I was wondering if there was a simpler way. But many thanks for your kind reply (as always). |
|
Dear Hai, import nglview as nv
v = nv.NGLWidget()
c0 = v.add_component('esp.mol')
c1 = v.add_component('esp.dx')
c1.clear()
c0.add_surface(
color_scheme='volume',
color_volume=c1,
colorScale='rwb',
colorDomain=[ -0.05, 0.05 ],
opacity=0.5
)
vHowever, with my initial example, a similar approach is still not working: view = nv.NGLWidget()
c0 = view.add_component('water_den.cube')
c1 = view.add_component('water_pot.cube')
c0.clear()
c0.add_surface(color_scheme='volume',
color_volume=c1,
colorScale='rwb',
colorDomain=[ -0.05, 0.05 ],
opacity=0.5
)
view
As a side point, if I load these same CUBE files into VMD (using a protocol similar to the VMD section here ), they do display fine. Any ideas? |
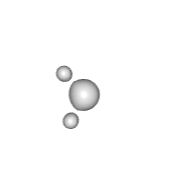
|
Thank you for the great tool. Is it possible to set opacity to residues at specific indices or a range of indices? I'm using the cartoon representation. Thanks! |
@mjw99 Can you please try NGL live example first by using your files? |
hi @jessevig, may be use multiple cartoon representations. Saying you have 5 residues and want to set opacity for some of them. |
|
@arose Does NGL support setting opacity for some residues in ONE representation? thanks. |
|
Is there a simple example for animating a protein structure to rotate? I found this but wasn't sure how to invoke it from the jupyter notebook: https://github.com/arose/ngl/blob/master/src/controls/animation-controls.ts |
|
@jessevig may be use |
|
I thought I tried that one before, but it didn't show an animation associated with spinning, it just changed the viewing angle. Is it supposed to show the animation? Thanks. |
|
@jessevig I see. Please use |
|
Thanks. Do you know if it's possible to display text sideways, i.e. rotated 90 degrees? |
I am not sure. May be playing with |
|
Hello, I've been trying to use ngl to visualize DNA oligos from a .lammpstrj trajectory file and .psf topology. I am able to load the data without any error, but when I visualize a frame I am not able to recover the bonding information contained in the psf file. I believe that some bonds are forming based off proximity (they look like random triangles) but these are not reflective of what I see in vmd for example. Here is the code I am using and the corresponding visualization: `frame = md.load_frame('../GC-core_321K_vis.lammpstrj', 0, top='../GC-core-ions.psf') view = nv.show_mdtraj(frame) strand_len = 29 view`
|

|
hi @mrjoness, Load test.pdb to VMD, do you still see the bond correctly? |
|
When I use test.pdb in vmd I don't see the correct bonds either... I guess there's a problem with the mdtraj method (or my psf file) and not necessarily nglviewer? Is there anything you recommend to fix this? |
|
@mrjoness Either:
|
|
Hi, [EDIT] More in general, what is the syntax for selecting a component and its atoms? I have a small molecule loaded as a separate component and I cannot understand how to find out the identifiers assigned to its atoms |
|
hi @luponzo86, I don't think cc @arose |
|
Thanks, @hainm I then tried to combine the two molecules together with OpenBabel: and it works, but then I don't know how to tell the two molecules apart during selection. Is there a way to figure out the identifiers of the two molecules? Is there something like Thanks! |
Can you try |
Thanks a lot, very useful! I was finally able to combine the two molecules ( |
|
@luponzo86 fantastic. |
|
Is there a way to display a view from inside a function in a Jupyter Notebook: cell 1: cell 2: output: The trajectory isn't displayed, but it works fine when I don't use a function. Is there a view1.show() type function I should use instead? |
|
hi @markperri, just |
|
@hainm thanks! |
|
hi @gmacari: sorry for the delay. There is no direct way to do that, however, you can use |
|
I have a problem with the size of the ngl view in the embedded viewer. The |
|
@jochym I don't know for now, let me think a bit. |
|
@jochym The best way currently is to wrap the
|

|
This is absolutely fine for my needs - probably not for everyone, but it is a perfectly acceptable solution. Thanks! |
|
Added to FAQ (#945) |
|
Hi, Is there a way to update the atomic species in the NGLViewer widget when changing frames? Currently it only updates the positions of the species, however I'm working on trajectories which has varying species as well. As a an example (from ASE): Here, changing the frames does not actually update the atomic species. |
hi @atygesen No, |
|
Hi, does anyone know how to get the periodic boundary conditions to show in the visuals? this is what my molecules look like: I want it to look something like this:
|

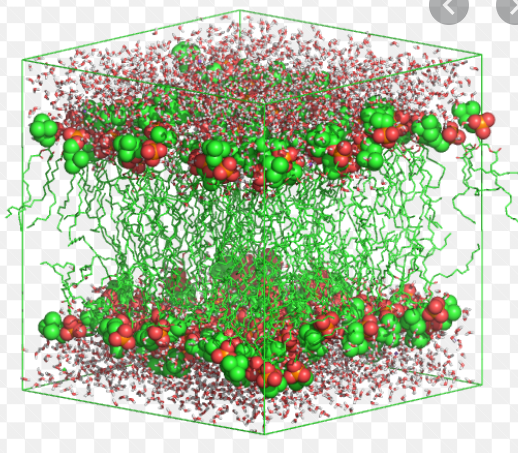
|
You need to have the periodic boundary conditions turned on in the structure (at least in the ase structures it works that way) and you need to |
thank you so much! |

|
@hainm Can you label this as |
Done. Thanks @jaimergp |
also, do you know if there is a way to add color to the unit cell's background? |
No. And you can not even change the color of the unitcell. |
|
Open new thread: #948 |
In case this may still interest anyone. The example actually works if the isolevel of the surface is specified: |
|
Thanks @jcerezochem |
|
hi |

i can't get the rendered picture ; |
|
Hi,
Replace “print(view)” by “view” to display the view first.
…On Fri, Oct 29, 2021 at 11:46 PM Hao-Yu Wang | 王昊宇 | Stefan < ***@***.***> wrote:
hi when i am using the nglview, i load the traj file and then want to show
the traj with nglview, i meet the question like this [image: 截屏2021-10-30
上午11 42 01]
<https://user-images.githubusercontent.com/75574822/139519100-5051c126-ecbd-492e-a74b-78c93b8afe2b.png>
:
i can't get the rendered picture ;
there is a picture which is can't load : (
—
You are receiving this because you modified the open/close state.
Reply to this email directly, view it on GitHub
<#901 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABB645JELZTV5ZHVVWHGZZ3UJNTBTANCNFSM4NJTLKXA>
.
Triage notifications on the go with GitHub Mobile for iOS
<https://apps.apple.com/app/apple-store/id1477376905?ct=notification-email&mt=8&pt=524675>
or Android
<https://play.google.com/store/apps/details?id=com.github.android&referrer=utm_campaign%3Dnotification-email%26utm_medium%3Demail%26utm_source%3Dgithub>.
|
|
hi
It works !
Thank you so much !
—
On 10/30/2021 21:47,Hai ***@***.***> wrote:
Hi,
Replace “print(view)” by “view” to display the view first.
On Fri, Oct 29, 2021 at 11:46 PM Hao-Yu Wang | 王昊宇 | Stefan <
***@***.***> wrote:
hi when i am using the nglview, i load the traj file and then want to show
the traj with nglview, i meet the question like this [image: 截屏2021-10-30
上午11 42 01]
<https://user-images.githubusercontent.com/75574822/139519100-5051c126-ecbd-492e-a74b-78c93b8afe2b.png>
:
i can't get the rendered picture ;
there is a picture which is can't load : (
—
You are receiving this because you modified the open/close state.
Reply to this email directly, view it on GitHub
<#901 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABB645JELZTV5ZHVVWHGZZ3UJNTBTANCNFSM4NJTLKXA>
.
Triage notifications on the go with GitHub Mobile for iOS
<https://apps.apple.com/app/apple-store/id1477376905?ct=notification-email&mt=8&pt=524675>
or Android
<https://play.google.com/store/apps/details?id=com.github.android&referrer=utm_campaign%3Dnotification-email%26utm_medium%3Demail%26utm_source%3Dgithub>.
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub, or unsubscribe.
Triage notifications on the go with GitHub Mobile for iOS or Android.
|
|
Hello Hai, I am super interested in the Shape method. I would like to draw a cylinder connecting two atoms on two different macromolecule residues. How can I retrieve the coordinates of these atoms? Once I have the atom index I can do that, but I want to be able to retrieve given residue numbers and chain IDs which is the data I have. Thanks! Sam
|
For any one interested; there is a demo of this here: https://github.com/mjw99/pyscf_mep |
|
Thanks @mjw99 |
|
I have a working installation of nglview using jupyter lab. My problem is i'd like to be able to "Restart Kernel and Run all Cells" and see the nglview as the cells are run, however, the views aren't displayed until after all the kernels finish or I stop the kernel. Is there anyway to get my desired behavior? Nglview (Version 1.1.7, from Bioconda) Conda environment yml name: biobb_amber
|
For any questions relating to how/why/ ... Or anything you don't know where to ask.
For bug, suggestion or anything you want to ask separately, please open seperate issue
PS:
The text was updated successfully, but these errors were encountered: