nscf-uniform calculation and data query in MgO band structure tutorial #283
Comments
|
Hi @zezhong-zhang, thanks for this very detailed issue. You are correct that this is due to use of A recent update to the I am looking into a fix for this now. |
|
The underlying issue is referenced in materialsproject/pymatgen#1453. |
|
Hi @utf Many thanks for bringing this issue into attention. I have checked the discussion, looking forward to the updated MPNonSCFSet. I think the data query should be a separate issue that I will edit soon. |
|
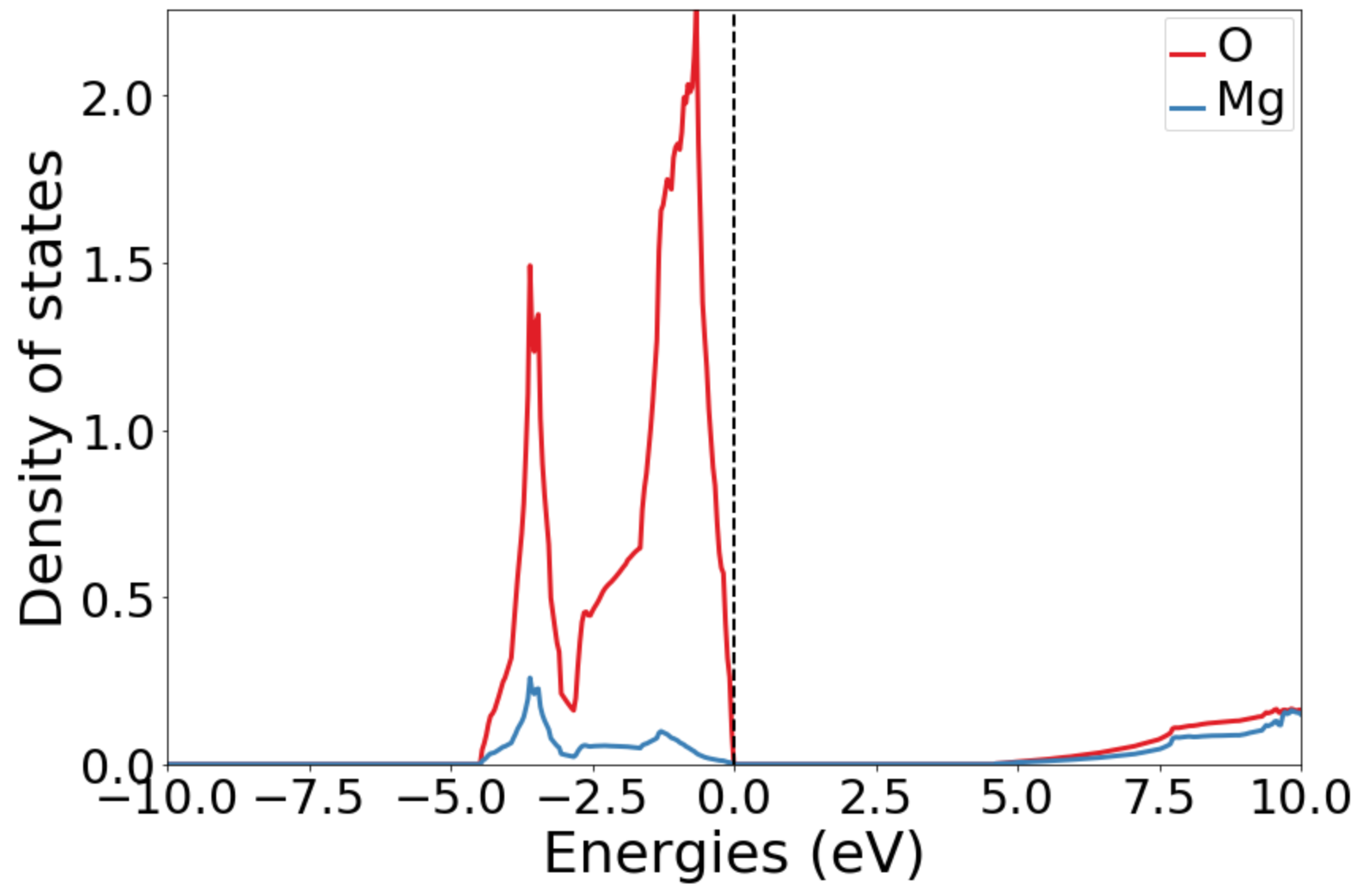
Hi @utf, I have another tested run after the update and the nscf-uniform calculation can now finish. However, the plotted DOS is different from the example, see as attached, while the bandstructure plots properly.
|

|
Here is the DOS from the tutorial for comparison. |
|
@zezhong-zhang Can you remove the
|
|
Hi @zezhong-zhang, we have updated the default density of states smearing parameter (ISMEAR) in the latest update of pymatgen. This accounts for the difference you see in the shape of the density of states. As you can see, the new density of states shows much more detail than the previous version, however, I am surprised that the intensity has increased so much. I will:
|
|
Hi @bocklund and @utf, here are the plots without ylimit. I also add the calcualted Fermi level and bandgap. I am really surprised by the significant difference. Fermi energy: 3.93249361 eV
Without xlimit. The peaks are vastly different from the Materials Project in the range between (-4 eV, 8eV) |



I was following the MgO band structure tutorial. The structure optimisation, statistic calculation and non-self-consistent-line calculations are fine. But for nscf-uniform calculation, vasp fails namelessly as shown in the fig and custodian cannot handle. As suggested by Steven on the google group, it may due to the kpoints generated not matching well with the tetrahedron method ISMEAR=-5.
The text was updated successfully, but these errors were encountered: