{kind=link}
Palshikar, M. G., Palli, R., Tyrell, A., Maggirwar, S., Schifitto, G., Singh, M. V., & Thakar, J. (2022). Executable models of immune signaling pathways in HIV-associated atherosclerosis. npj Systems Biology and Applications, 8(1), 35. doi:10.1038/s41540-022-00246
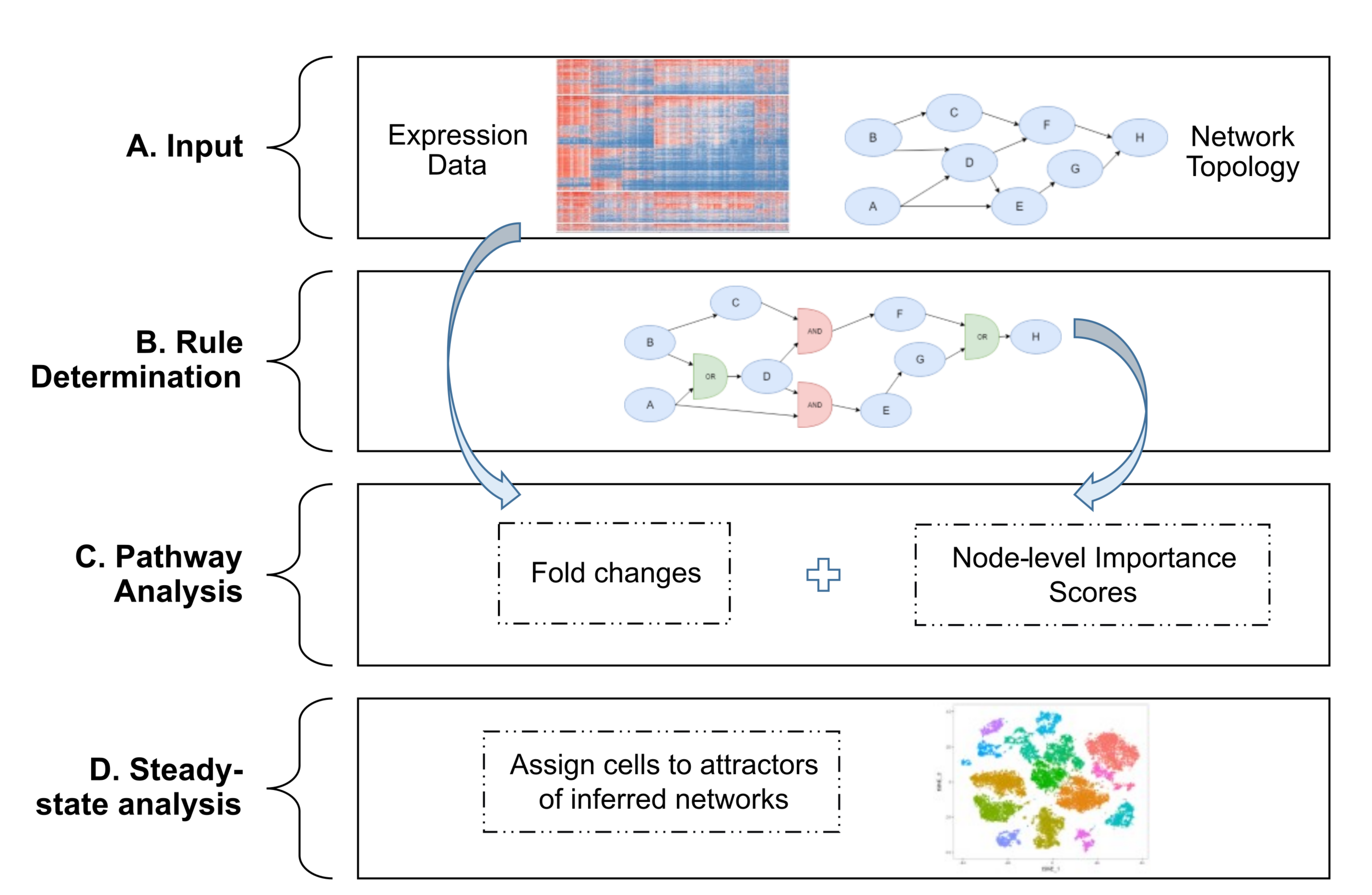
To use single-cellRNA seq data to develop executable models of signaling pathways that drive cellular states, we developed the single-cell Boolean Omics Network Invariant Time Analysis (scBONITA) algorithm. ScBONITA
- uses scRNA-seq data to infer Boolean regulatory rules for topologically characterized networks
- prioritizes genes based on their impact on signaling
- performs pathway analysis, and
- maps sequenced cells to characteristic signaling states of these networks
We show in our manuscript that scBONITA learns dynamic models of signaling pathways to identify pathways significantly dysregulated in HIV-associated atherosclerosis. These pathways indicate that cell migration into the vascular endothelium is significantly affected by dysregulated lipid signaling in AS+ PLWH. Dynamic modeling facilitates pathway-based characterization of cellular states that are not apparent in gene expression analyses.
This repository includes code for preliminary analysis of our HIV/AS scRNA-seq dataset GSE198339
- Link to dataset: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198339
- Link to analysis code: https://github.com/Thakar-Lab/scBONITA/tree/main/HIVAS_dataset_preliminary_analysis
single-cell RNA sequencing; Boolean networks; HIV; atherosclerosis; pathway analysis; network modeling
[email protected]; [email protected]
https://github.com/Thakar-Lab/scBONITA/issues
scBONITA requires Python 3.6 or newer.
scBONITA is currently designed to be run on SLURM systems, with minimally complicated transfer to other distributed computing systems.
We recommend that the scBONITA pipeline is used on a high-performance system with adequate computational resources. We find that the amount of RAM required for large scRNA-seq datasets is in excess of 10G; we hence don't recommend running rule inference and attractor analysis on a desktop computer.
-
Download a zipped folder from github.com/Thakar-Lab/scBONITA, OR
-
Use git to clone this repository
git clone https://github.com/YOUR-USERNAME/YOUR-REPOSITORY
Modify the C file to reflect the size of the training data set - line 7: NODE is the number of nodes in the dataset (or larger) - line 8: CELL is the number of cells to be sampled from the dataset (or larger)
Next, the C code must be compiled using the make file. Navigate to the folder in which the scBONITA package is located and in a Bash terminal, type
make
Install the scBonita conda environment from the provided yml file OR the provided spec-file (PREFERRED), and activate the conda environment. Type the following commands into a terminal. Note - you should be in the directory containing the spec-file/yml file.
module load anaconda3/2020.07
conda create --name scBonita --file spec-file.txt
source activate scBonita
If there are errors at later stages suggesting that packages are not installed/dependencies are not installed or need to be updated, you will need to install packages into this conda environment. I suggest googling 'install package xyz in conda' and following the instructions.
Please refer to the tutorial "Rule_Inference_With_scBONITA.ipynb" for a demonstration of rule inference with scBONITA using a test dataset packaged with scBONITA and a list of parameters for the setup. More information can also be found by typing python3 pipeline.py --help into the terminal.
Copy the following files into the src/scBonita folder that you downloaded from github:
- scBONITA needs a training dataset in matrix-style format; this is usually a tab or comma-delimited file with columns as cells and rows as features. The first column should be feature names and the first row should be cell IDs. The units of the expression data will typically be a variant of log2(TPM +1). The first column should be labeled 'Genes' or similar. The first row should be cell IDs. The cell IDs should be the same as the cell IDs in the metadata file (see below).
| Genes | Cell1 | Cell2 | Cell3 | Cell4 |
|---|---|---|---|---|
| Gene1 | 1.1 | 2.1 | . | . |
| Gene2 | 1.2 | 2.3 | . | . |
| Gene3 | 1.3 | . | . | . |
| Gene4 | 1.4 | . | . | . |
- metadata (or conditions) in a matrix format, where rows are cells and columns are cell properties/treatment/condition/etc. Entries must be 1s or 0s.
Example for dataset containing a mixture of different types of monocytes and B cells:
| CellID | CD14_monocyte | CD16_monocyte | Memory_Bcell | Naive_Bcell |
|---|---|---|---|---|
| Cell1 | 1 | 0 | 0 | 0 |
| Cell2 | 0 | 0 | 1 | 0 |
| Cell3 | 0 | 1 | 0 | 0 |
| Cell4 | 0 | 0 | 0 | 1 |
- a contrast file. This is similar to a design matrix. Each row contains the pair of conditions that need to be compared, separated by a tab. The conditions should be columns from the metadata file. For example,
Example if you wanted to compare cell types as defined in the metadata example above:
| CD14_monocyte | CD16_monocyte |
| Memory_Bcell | Naive_Bcell |
The following command runs scBONITA setup for a 20000*10000 comma-separated data set "example.csv". It downloads the KEGG pathways hsa00010 and hsa00020 for rule inference.
python3.6 pipeline.py --dataFile "example.csv" --fullPipeline 1 --maxNodes 20000 --separator "," --listOfKEGGPathways "00010" "00020" --getKEGGPathways 1 --organism hsa cvThreshold None
Step 2: Rule inference and calculation of node importance score for the networks specified in Step 1 (setup).
Step 1 generates sbatch files that enter the specified slurm queue. In a typical use case, these jobs should execute automatically. We recommend that users periodically check the slurm queue and the log files.
The pathway analysis script has the following arguments:
-
dataFile
Specify the name of the file containing processed scRNA-seq data
-
conditions
Specify the name of the file containing cell-wise condition labels, ie, metadata for each cell in the training dataset. The columns are condition variables and the rows are cells. The first column must be cell names that correspond to the columns in the training data file. The column names must contain the variables specified in the contrast file (see contrast --help for more information).
-
contrast
A text file where each line contains the two conditions (corresponding to column labels in the conditions file) are to be compared during pathway analysis.
-
conditions_separator
Separator for the conditions file. Must be one of 'comma', 'space' or 'tab' (spell out words, escape characters will not work).",
-
contrast_separator
Separator for the contrast file. Must be one of 'comma', 'space' or 'tab' (spell out words, escape characters will not work).",
-
pathwayList
Paths to GRAPHML files that should be used for scBONITA analysis. Usually networks from non-KEGG sources, saved in GRAPHML format. The default value is to use sll the networks initially used for rule inference.
Example usage with the provided example files in the data folder:
python3.6 pathwayAnalysis.py --dataFile "data/trainingData.csv" --conditions "data/conditions.txt" --contrast "data/contrast.txt" --conditions_separator "comma" --contrast_separator "comma" --pathwayList "hsa00010"
Please refer to the tutorial "Pathway_Analysis_With_scBONITA.ipynb" for suggestions on analysis of the output of scBONITA PA
Please refer to the tutorial "Attractor_Analysis_With_scBONITA.ipynb" for a demonstration of attractor analysis with scBONITA using a test dataset packaged with scBONITA