Introduction
Explicit networks have a biological interpretation:
internal nodes represent ancestral species (or populations);
the main evolutionary history is depicted by the "major tree".
Various methods that estimate explicit networks use models
that account for ILS and for gene tree estimation error.
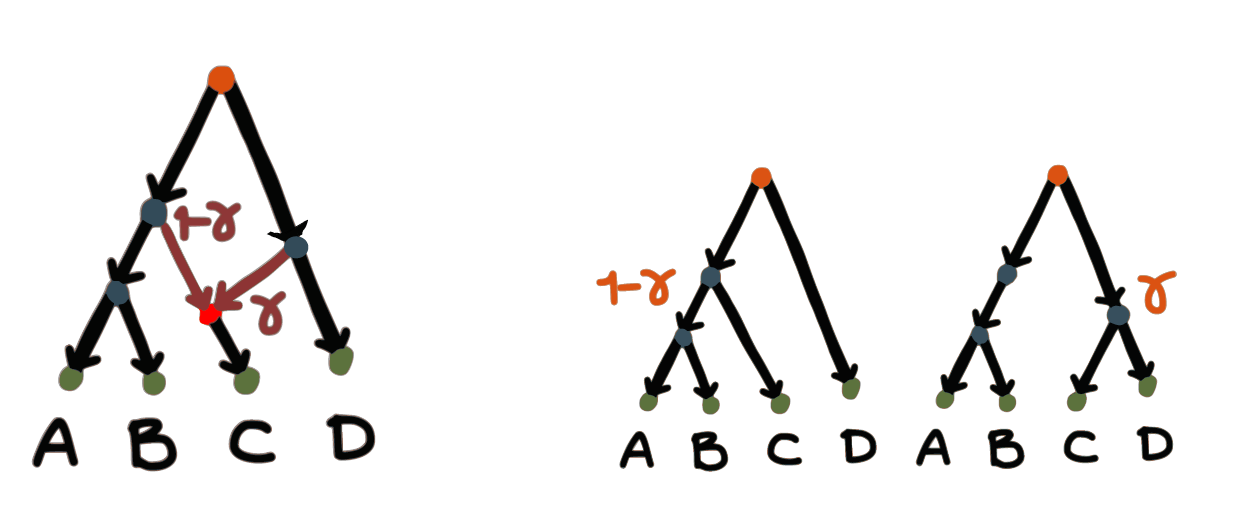
Implicit networks are typically descriptive:
internal nodes do not represent ancestral species.
Implicit networks do not discriminate between ILS,
gene flow/hybridization or gene tree estimation error,
and can be hard to interpret biologically.

Recall that in parenthetical format, internal nodes can have a name:
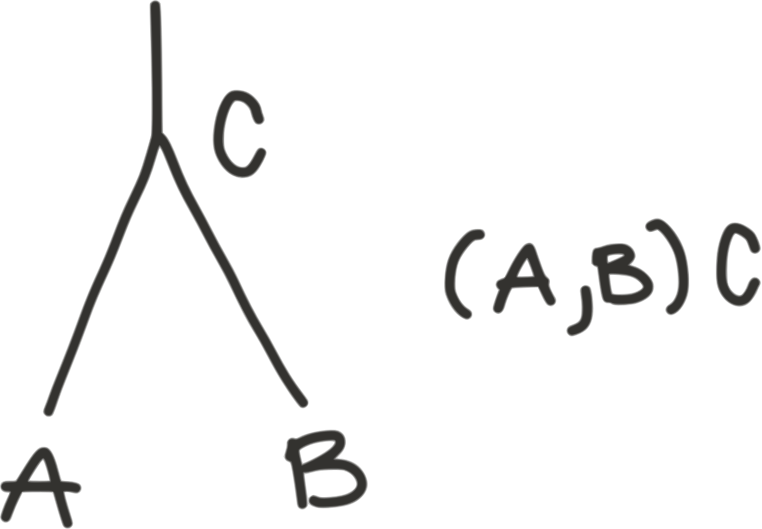
To represent networks in parenthetical format, we simply need to split
each hybrid node into two nodes with the same name:

By convention, the hybrid tag is # + H,LGT,R + number, and the minor
hybrid edge leads to a leaf.
Thus, we get: (((A,(B)#H1),(C,#H1)),D);. We can write inheritance
probabilities in the parenthetical format: (C,#H1):branch length:bootstrap support:inheritance probability.
In Julia:
using PhyloNetworks # needed in each new julia session. the first time takes a while
using PhyloPlots
net = readTopology("(((A,(B)#H1:::0.9),(C,#H1:::0.1)),D);")
writeTopology(net)net, as obtained above, is a Julia object that can be manipulated inside Julia. For
example, we can plot it with:
plot(net, :R);This should open up a new window containing the plot. Do not close this plot window! Future plots will need to use the same window.
This new window might not appear on a remote cluster, if windows are not "forwarded"
from the remote machine to the local machine (X or X11 forwarding).
A first option is to log out of the machine and ssh back into that remote machine
using the -X option, like: ssh -X [email protected].
A second alternative is to create a pdf (for example) with the plot, and then go fetch that pdf file:
using RCall
R"pdf"("plot1.pdf", width=3, height=3);
plot(net, :R);
R"dev.off"();To see that a pdf file was created, a first step can be to type ;
at the julia prompt: this changes the prompt to a shell prompt,
and then we can type any shell command, such as ls or ls plot*
to see if our file was indeed created, or some other command
to open it or transfer it back.
The hybrid edges are displayed in blue: dark blue for the major edge,
and light blue for the minor edge.
There are options to show node/edge numbers, edge length, inheritance probabilities,
change colors, etc. Type ? within Julia to turn on the help mode, and then plot
to get help and options on the plot function.
We can also change the root placement to a different edge or node, or to an outgroup.
rootatnode!(net,"A")
# R"pdf"("plot2.pdf", width=3, height=3) # uncomment to save as pdf
R"par"(mar=[0.5,0.5,0.5,0.5]); # to reduce margins in next plot
plot(net, :R, showEdgeNumber=true)
# R"dev.off"()Warning: we need to be careful with the placement of the root, because it should agree with the direction of the hybrid edges.
PhyloNetworks is a Julia package for explicit phylogenetic networks with functions to
- read/write networks in parenthetical format
- plot networks
- change the root, or make other network modifications
- estimate the maximum pseudolikelihood network
- do a bootstrap analysis, summarize a sample of (bootstrap) networks
- comparative methods for trait evolution on a network
Julia is a high-level and interactive programming language (like R or Matlab), but it is also high-performance (like C). Also, Julia allows for collaboration in an easy way through Github. See here to install Julia, then refer to the PhyloNetworks documentation to install and use PhyloNetworks.
To change the directory used by julia in a session (warning: you do not need to do this for the MBL workshop), say to a folder named "tutorial" in your "Desktop", you have 2 options.
- quit your session, navigate to the directory and restart julia there.
- or change the working directory within your Julia session like this
homedir() # just to show the command homedir() to extract the path to your home directory
cd("$(homedir())/PhyloNetworks.jl.wiki/data_results/baseline.gamma0.3_n30") # changes the working directory
pwd() # just to check: print working directoryif you clone the wiki repository, or
homedir() # just to show the command homedir() to extract the path to your home directory
cd("$(homedir())/moledata/phylo-networks/data_results/baseline.gamma0.3_n30") # changes the working directory
pwd() # just to check: print working directoryif you copied and pasted from the MOLE data folder (for MBL workshop).
If you are going to use the example data, make sure you are inside the
folder baseline.gamma0.3_n30. If you are using your own data, make
sure that you are in the folder where the data are.
Next: estimate a network from data with SNaQ.