DeeReCT-TSS: A novel meta-learning-based method annotates TSS in multiple cell types based on DNA sequences and RNA-seq data
This repository contains the implementation of DeeReCT-TSS from
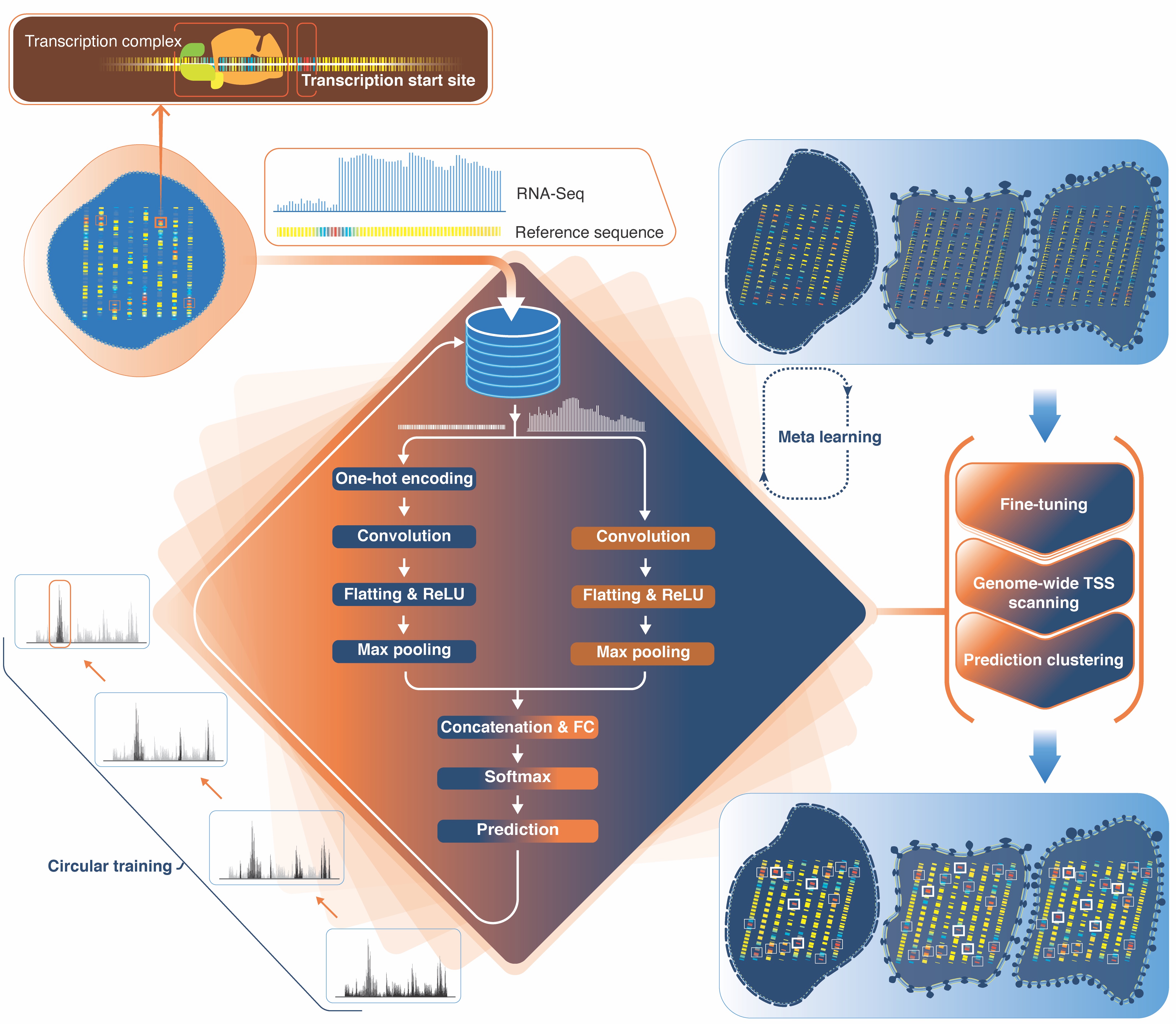
Juexiao Zhou, Bin Zhang, et al. "Annotating TSSs in Multiple Cell Types Based on DNA Sequence and RNA-seq Data via DeeReCT-TSS"
If you use our work in your research, please consider cite our paper:
@article{zhou2022annotating,
title={Annotating TSSs in Multiple Cell Types Based on DNA Sequence and RNA-seq Data via DeeReCT-TSS},
author={Zhou, Juexiao and Zhang, Bin and Li, Haoyang and Zhou, Longxi and Li, Zhongxiao and Long, Yongkang and Han, Wenkai and Wang, Mengran and Cui, Huanhuan and Li, Jingjing and others},
journal={Genomics, Proteomics \& Bioinformatics},
year={2022},
publisher={Elsevier}
}
The code is tested with the following dependencies:
-
python=3.6
-
biopython=1.78
-
bedtools=2.30.0
-
cudatoolkit=10.1.243
-
cudnn=7.6.5
-
numpy=1.19.2
-
scipy=1.5.2
-
pandas=1.1.3
-
scipy=1.5.2
-
scikit-learn 0.22.1
-
tensorflow-gpu=1.14.0
-
Seaborn 0.11.1
-
matplotlib=3.3.4
-
seaborn=0.11.1
-
samtools
The code is not guaranteed to work if different versions are used.
To analyze bam files with a size around 10G, each thread requires 4-5G memory when the job is splitted into 25 threads.
bash ./run.sh \
path/to/Aligned.sortedByCoord.out.bam \ #(the aligned RNA-Seq bam file)
path/to/gencode.v38.pcg.extups5k.bed \ #(regions for scanning, a example file of all protein coding genes is provided under the folder /ref)
path/to/model.npz \ #(the pre-trained models are provided under the folder /model)
path/to/reference_genome.fa \ #(reference genome sequencing in the "FASTA" format, a example file is provided under the folder /ref)
path/to/output \
0/1/2 ifstranded \
25 #(number of threads)
eg:
bash ./run.sh \
../DeeReCT-TSS_release/data/TCGA-AA-3517-11A-01R-A32Z-07/rnaseq/Aligned.sortedByCoord.out.bam \
../DeeReCT-TSS_release/ref/gencode.v38.pcg.extups5k.bed \
../DeeReCT-TSS_release/model/colon_model/model_best.npz \
../DeeReCT-TSS_release/ref/hg38/hg38.fa \
./test_out/ \
0 \
25
The reference genome file can be download from "https://www.gencodegenes.org" or other database. i.e. Ensembl, UCSC and NCBI.
The file marking the regions for scanning should be in "BED" format. A simple way to generate the file for scanning all protein coding genes is shown below:
1, Download gene annotation (gtf file) from "http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_38/gencode.v38.annotation.gtf.gz"
2, Select the rows including gene information, filter out the protein coding genes, extend 5kb from the gene start and convert to "BED" format
zcat gencode.v38.annotation.gtf.gz | awk '$3 == "gene"' | grep "protein_coding" | awk '{OFS="\t"} {if($6 == "+") print $1,$4-5000,$5,$10":"$14,$12,$7; else print $1,$4,$5+5000,$10":"$14,$12,$7}' | sed s/[\"\;]//g > gencode.v38.pcg.extups5k.bed
There will be two output files under the output directory: "combined.raw.prediction" and "combined.predicted.cluster"
The former one is the raw prediction score in a extended bedgraph format from the deep learning model. Column 1-3 indicate the genomic coordiante, column 4 is the predcition score, and column 5 is the strand.
The later one is the final predicted TSS after clustering the raw prediction score in a extended bed format. Column 1-3 indicate the TSS loci, column 4 is the gene where the predicted TSS is associated, column 5 is the clustered prediction score, column 6 is the strand, and column 7 is a empirical P value.
This project is supported by KAUST and SUSTech.