diff --git a/cell_type_annotation.md b/cell_type_annotation.md
index b91e30f..050af66 100644
--- a/cell_type_annotation.md
+++ b/cell_type_annotation.md
@@ -8,8 +8,8 @@ editor_options:
---
::: questions
-- How to identify groups of cells with similar expression profiles?
-- How to identify genes that drive separation between these groups of cells?
+- How can we identify groups of cells with similar expression profiles?
+- How can we identify genes that drive separation between these groups of cells?
- How to leverage reference datasets and known marker genes for the cell type annotation of new datasets?
:::
@@ -36,6 +36,8 @@ library(scran)
## Data retrieval
+We'll be using the same set of WT chimeric mouse embryo data:
+
``` r
sce <- WTChimeraData(samples = 5, type = "processed")
@@ -57,7 +59,7 @@ mainExpName: NULL
altExpNames(0):
```
-To speed up the computations, we subsample the dataset to 1,000 cells.
+To speed up the computations, we take a random subset of 1,000 cells.
``` r
@@ -68,6 +70,8 @@ sce <- sce[,ind]
## Preprocessing
+The SCE object needs to contain log-normalized expression counts as well as PCA coordinates in the reduced dimensions, so we compute those here:
+
``` r
sce <- logNormCounts(sce)
@@ -105,12 +109,14 @@ algorithm](https://doi.org/10.1088/1742-5468/2008/10/P10008) for
community detection. All calculations are performed using the top PCs to
take advantage of data compression and denoising. This function returns
a vector containing cluster assignments for each cell in our
-`SingleCellExperiment` object.
+`SingleCellExperiment` object. We use the `colLabels()` function to assign the
+cluster labels as a factor in the column data.
``` r
colLabels(sce) <- clusterCells(sce, use.dimred = "PCA",
BLUSPARAM = NNGraphParam(cluster.fun = "louvain"))
+
table(colLabels(sce))
```
@@ -120,19 +126,29 @@ table(colLabels(sce))
100 160 99 141 63 93 60 108 44 91 41
```
-We assign the cluster assignments back into our `SingleCellExperiment`
-object as a `factor` in the column metadata. This allows us to
-conveniently visualize the distribution of clusters in eg. a *t*-SNE or
-a UMAP.
+We can now overlay the cluster labels as color on a UMAP plot:
``` r
sce <- runUMAP(sce, dimred = "PCA")
+
plotReducedDim(sce, "UMAP", color_by = "label")
```
 +:::: challenge
+
+Our clusters look semi-reasonable, but what if we wanted to make them less granular? Look at the help documentation for `?clusterCells` and `?NNGraphParam` to find out what we'd need to change to get fewer, larger clusters.
+
+::: solution
+
+We see in the help documentation for `?clusterCells` that all of the clustering algorithm details are handled through the `BLUSPARAM` argument, which needs to provide a `BlusterParam` object (of which `NNGraphParam` is a sub-class). Each type of clustering algorithm will have some sort of hyper-parameter that controls the granularity of the output clusters. Looking at `?NNGraphParam` specifically, we see an argument called `k` which is described as "An integer scalar specifying the number of nearest neighbors to consider during graph construction." If the clustering process has to connect larger sets of neighbors, the graph will tend to be cut into larger groups, resulting in less granular clusters. Try the two code blocks above once more with `k = 20`. Given their visual differences, do you think one set of clusters is "right" and the other is "wrong"?
+:::
+
+::::
+
+
## Marker gene detection
To interpret clustering results as obtained in the previous section, we
@@ -147,7 +163,7 @@ testing for differential expression between clusters. If a gene is
strongly DE between clusters, it is likely to have driven the separation
of cells in the clustering algorithm.
-Here, we perform a Wilcoxon rank sum test against a log2 fold change
+Here, we use `findMarkers()` to perform a Wilcoxon rank sum test against a log2 fold change
threshold of 1, focusing on up-regulated (positive) markers in one
cluster when compared to another cluster.
@@ -163,6 +179,8 @@ List of length 11
names(11): 1 2 3 4 5 6 7 8 9 10 11
```
+
+
The resulting object contains a sorted marker gene list for each
cluster, in which the top genes are those that contribute the most to
the separation of that cluster from mall other clusters.
@@ -238,6 +256,22 @@ plotExpression(sce, features = top.markers, x = "label", color_by = "label")
+:::: challenge
+
+Our clusters look semi-reasonable, but what if we wanted to make them less granular? Look at the help documentation for `?clusterCells` and `?NNGraphParam` to find out what we'd need to change to get fewer, larger clusters.
+
+::: solution
+
+We see in the help documentation for `?clusterCells` that all of the clustering algorithm details are handled through the `BLUSPARAM` argument, which needs to provide a `BlusterParam` object (of which `NNGraphParam` is a sub-class). Each type of clustering algorithm will have some sort of hyper-parameter that controls the granularity of the output clusters. Looking at `?NNGraphParam` specifically, we see an argument called `k` which is described as "An integer scalar specifying the number of nearest neighbors to consider during graph construction." If the clustering process has to connect larger sets of neighbors, the graph will tend to be cut into larger groups, resulting in less granular clusters. Try the two code blocks above once more with `k = 20`. Given their visual differences, do you think one set of clusters is "right" and the other is "wrong"?
+:::
+
+::::
+
+
## Marker gene detection
To interpret clustering results as obtained in the previous section, we
@@ -147,7 +163,7 @@ testing for differential expression between clusters. If a gene is
strongly DE between clusters, it is likely to have driven the separation
of cells in the clustering algorithm.
-Here, we perform a Wilcoxon rank sum test against a log2 fold change
+Here, we use `findMarkers()` to perform a Wilcoxon rank sum test against a log2 fold change
threshold of 1, focusing on up-regulated (positive) markers in one
cluster when compared to another cluster.
@@ -163,6 +179,8 @@ List of length 11
names(11): 1 2 3 4 5 6 7 8 9 10 11
```
+
+
The resulting object contains a sorted marker gene list for each
cluster, in which the top genes are those that contribute the most to
the separation of that cluster from mall other clusters.
@@ -238,6 +256,22 @@ plotExpression(sce, features = top.markers, x = "label", color_by = "label")
 +Clearly, not every marker gene distinguishes cluster 1 from every other cluster. However, with a combination of multiple marker genes it's possible to clearly identify gene patterns that are unique to cluster 1. It's sort of like the 20 questions game - with answers to the right questions about a cell (e.g. "Do you highly express Ptn?"), you can clearly identify what cluster it falls in.
+
+:::: challenge
+
+Why do you think marker genes are found by aggregating pairwise comparisons rather than iteratively comparing each cluster to all other clusters?
+
+::: solution
+
+One important reason why is because averages over all other clusters can be sensitive to the cell type composition. If a rare cell type shows up in one sample, the most discriminative marker genes found in this way could be very different from those found in another sample where the rare cell type is absent.
+
+Generally, it's good to keep in mind that the concept of "everything else" is not a stable basis for comparison. Read that sentence again, because its a subtle but broadly applicable point. Think about it and you can probably identify analogous issues in fields outside of single-cell analysis. It frequently comes up when comparisons between multiple categories are involved.
+
+:::
+::::
+
+
## Cell type annotation
The most challenging task in scRNA-seq data analysis is arguably the
@@ -332,6 +366,8 @@ ind <- sample(ncol(ref), 1000)
ref <- ref[,ind]
```
+You can see we have an assortment of different cell types in the reference (with varying frequency):
+
``` r
tab <- sort(table(ref$celltype), decreasing = TRUE)
@@ -411,9 +447,13 @@ sce.mat <- as.matrix(assay(sce, "logcounts"))
ref.mat <- as.matrix(assay(ref, "logcounts"))
```
+Finally, run SingleR with the query and reference datasets:
+
``` r
-res <- SingleR(test = sce.mat, ref = ref.mat, labels = ref$celltype)
+res <- SingleR(test = sce.mat,
+ ref = ref.mat,
+ labels = ref$celltype)
res
```
@@ -450,9 +490,7 @@ cell_11860 Forebrain/Midbrain/H..
We inspect the results using a heatmap of the per-cell and label scores.
Ideally, each cell should exhibit a high score in one label relative to
all of the others, indicating that the assignment to that label was
-unambiguous. This is largely the case for mesenchyme and endothelial
-cells, whereas we see expectedly more ambiguity between the two
-erythroid cell populations.
+unambiguous.
``` r
@@ -461,13 +499,17 @@ plotScoreHeatmap(res)
+Clearly, not every marker gene distinguishes cluster 1 from every other cluster. However, with a combination of multiple marker genes it's possible to clearly identify gene patterns that are unique to cluster 1. It's sort of like the 20 questions game - with answers to the right questions about a cell (e.g. "Do you highly express Ptn?"), you can clearly identify what cluster it falls in.
+
+:::: challenge
+
+Why do you think marker genes are found by aggregating pairwise comparisons rather than iteratively comparing each cluster to all other clusters?
+
+::: solution
+
+One important reason why is because averages over all other clusters can be sensitive to the cell type composition. If a rare cell type shows up in one sample, the most discriminative marker genes found in this way could be very different from those found in another sample where the rare cell type is absent.
+
+Generally, it's good to keep in mind that the concept of "everything else" is not a stable basis for comparison. Read that sentence again, because its a subtle but broadly applicable point. Think about it and you can probably identify analogous issues in fields outside of single-cell analysis. It frequently comes up when comparisons between multiple categories are involved.
+
+:::
+::::
+
+
## Cell type annotation
The most challenging task in scRNA-seq data analysis is arguably the
@@ -332,6 +366,8 @@ ind <- sample(ncol(ref), 1000)
ref <- ref[,ind]
```
+You can see we have an assortment of different cell types in the reference (with varying frequency):
+
``` r
tab <- sort(table(ref$celltype), decreasing = TRUE)
@@ -411,9 +447,13 @@ sce.mat <- as.matrix(assay(sce, "logcounts"))
ref.mat <- as.matrix(assay(ref, "logcounts"))
```
+Finally, run SingleR with the query and reference datasets:
+
``` r
-res <- SingleR(test = sce.mat, ref = ref.mat, labels = ref$celltype)
+res <- SingleR(test = sce.mat,
+ ref = ref.mat,
+ labels = ref$celltype)
res
```
@@ -450,9 +490,7 @@ cell_11860 Forebrain/Midbrain/H..
We inspect the results using a heatmap of the per-cell and label scores.
Ideally, each cell should exhibit a high score in one label relative to
all of the others, indicating that the assignment to that label was
-unambiguous. This is largely the case for mesenchyme and endothelial
-cells, whereas we see expectedly more ambiguity between the two
-erythroid cell populations.
+unambiguous.
``` r
@@ -461,13 +499,17 @@ plotScoreHeatmap(res)
 -We also compare the cell type assignments with the clustering results to
-determine the identity of each cluster. Here, several cell type classes
-are nested within the same cluster, indicating that these clusters are
-composed of several transcriptomically similar cell populations (such as
-cluster 4 and 6). On the other hand, there are also instances where we
-have several clusters for the same cell type, indicating that the
-clustering represents finer subdivisions within these cell types.
+We obtained fairly unambiguous predictions for mesenchyme and endothelial
+cells, whereas we see expectedly more ambiguity between the two
+erythroid cell populations.
+
+We can also compare the cell type assignments with the unsupervised clustering
+results to determine the identity of each cluster. Here, several cell type
+classes are nested within the same cluster, indicating that these clusters are
+composed of several transcriptomically similar cell populations. On the other
+hand, there are also instances where we have several clusters for the same cell
+type, indicating that the clustering represents finer subdivisions within these
+cell types.
``` r
@@ -492,6 +534,32 @@ pheatmap(log2(tab + 10), color = colorRampPalette(c("white", "blue"))(101))
-We also compare the cell type assignments with the clustering results to
-determine the identity of each cluster. Here, several cell type classes
-are nested within the same cluster, indicating that these clusters are
-composed of several transcriptomically similar cell populations (such as
-cluster 4 and 6). On the other hand, there are also instances where we
-have several clusters for the same cell type, indicating that the
-clustering represents finer subdivisions within these cell types.
+We obtained fairly unambiguous predictions for mesenchyme and endothelial
+cells, whereas we see expectedly more ambiguity between the two
+erythroid cell populations.
+
+We can also compare the cell type assignments with the unsupervised clustering
+results to determine the identity of each cluster. Here, several cell type
+classes are nested within the same cluster, indicating that these clusters are
+composed of several transcriptomically similar cell populations. On the other
+hand, there are also instances where we have several clusters for the same cell
+type, indicating that the clustering represents finer subdivisions within these
+cell types.
``` r
@@ -492,6 +534,32 @@ pheatmap(log2(tab + 10), color = colorRampPalette(c("white", "blue"))(101))
 +:::: challenge
+
+SingleR can be computationally expensive. How do you set it to run in parallel?
+
+::: solution
+
+Use `BiocParallel` and the `BPPARAM` argument! This example will set it to use four cores on your laptop, but you can also configure BiocParallel to use cluster jobs.
+
+
+``` r
+library(BiocParallel)
+
+my_bpparam = MulticoreParam(workers = 4)
+
+res <- SingleR(test = sce.mat,
+ ref = ref.mat,
+ labels = ref$celltype,
+ BPPARAM = my_bpparam)
+```
+
+`BiocParallel` is the most common way to enable parallel computation in Bioconductor packages, so you can expect to see it elsewhere outside of SingleR.
+
+:::
+
+::::
+
### Assigning cell labels from gene sets
A related strategy is to explicitly identify sets of marker genes that
@@ -904,6 +972,19 @@ mixture, and the grey curve represents a fitted normal distribution.
Vertical lines represent threshold estimates corresponding to each
estimate of the distribution.
+:::: challenge
+
+The diagnostics don't look so good for some of the examples here. Which ones? Why?
+
+::: solution
+
+The example that jumps out most strongly to the eye is ExE endoderm, which doesn't show clear separate modes. Simultaneously, Endothelium seems to have three or four modes.
+
+Remember, this is an exploratory diagnostic, not the final word! At this point it'd be good to engage in some critical inspection of the results. Maybe we don't have enough / the best marker genes. In this particular case, the fact that we subsetted the reference set to 1000 cells probably didn't help.
+:::
+
+::::
+
## Session Info
diff --git a/eda_qc.md b/eda_qc.md
index bc81f47..7a9a9c3 100644
--- a/eda_qc.md
+++ b/eda_qc.md
@@ -72,18 +72,25 @@ From the experiment, we expect to have only a few thousand cells, while we can s
``` r
library(DropletUtils)
+library(ggplot2)
+
bcrank <- barcodeRanks(counts(sce))
# Only showing unique points for plotting speed.
uniq <- !duplicated(bcrank$rank)
-plot(bcrank$rank[uniq], bcrank$total[uniq], log="xy",
- xlab="Rank", ylab="Total UMI count", cex.lab=1.2)
-abline(h=metadata(bcrank)$inflection, col="darkgreen", lty=2)
-abline(h=metadata(bcrank)$knee, col="dodgerblue", lty=2)
+line_df = data.frame(cutoff = names(metadata(bcrank)),
+ value = unlist(metadata(bcrank)))
-legend("bottomleft", legend=c("Inflection", "Knee"),
- col=c("darkgreen", "dodgerblue"), lty=2, cex=1.2)
+ggplot(bcrank[uniq,], aes(rank, total)) +
+ geom_point() +
+ geom_hline(data = line_df,
+ aes(color = cutoff,
+ yintercept = value),
+ lty = 2) +
+ scale_x_log10() +
+ scale_y_log10() +
+ labs(y = "Total UMI count")
```
+:::: challenge
+
+SingleR can be computationally expensive. How do you set it to run in parallel?
+
+::: solution
+
+Use `BiocParallel` and the `BPPARAM` argument! This example will set it to use four cores on your laptop, but you can also configure BiocParallel to use cluster jobs.
+
+
+``` r
+library(BiocParallel)
+
+my_bpparam = MulticoreParam(workers = 4)
+
+res <- SingleR(test = sce.mat,
+ ref = ref.mat,
+ labels = ref$celltype,
+ BPPARAM = my_bpparam)
+```
+
+`BiocParallel` is the most common way to enable parallel computation in Bioconductor packages, so you can expect to see it elsewhere outside of SingleR.
+
+:::
+
+::::
+
### Assigning cell labels from gene sets
A related strategy is to explicitly identify sets of marker genes that
@@ -904,6 +972,19 @@ mixture, and the grey curve represents a fitted normal distribution.
Vertical lines represent threshold estimates corresponding to each
estimate of the distribution.
+:::: challenge
+
+The diagnostics don't look so good for some of the examples here. Which ones? Why?
+
+::: solution
+
+The example that jumps out most strongly to the eye is ExE endoderm, which doesn't show clear separate modes. Simultaneously, Endothelium seems to have three or four modes.
+
+Remember, this is an exploratory diagnostic, not the final word! At this point it'd be good to engage in some critical inspection of the results. Maybe we don't have enough / the best marker genes. In this particular case, the fact that we subsetted the reference set to 1000 cells probably didn't help.
+:::
+
+::::
+
## Session Info
diff --git a/eda_qc.md b/eda_qc.md
index bc81f47..7a9a9c3 100644
--- a/eda_qc.md
+++ b/eda_qc.md
@@ -72,18 +72,25 @@ From the experiment, we expect to have only a few thousand cells, while we can s
``` r
library(DropletUtils)
+library(ggplot2)
+
bcrank <- barcodeRanks(counts(sce))
# Only showing unique points for plotting speed.
uniq <- !duplicated(bcrank$rank)
-plot(bcrank$rank[uniq], bcrank$total[uniq], log="xy",
- xlab="Rank", ylab="Total UMI count", cex.lab=1.2)
-abline(h=metadata(bcrank)$inflection, col="darkgreen", lty=2)
-abline(h=metadata(bcrank)$knee, col="dodgerblue", lty=2)
+line_df = data.frame(cutoff = names(metadata(bcrank)),
+ value = unlist(metadata(bcrank)))
-legend("bottomleft", legend=c("Inflection", "Knee"),
- col=c("darkgreen", "dodgerblue"), lty=2, cex=1.2)
+ggplot(bcrank[uniq,], aes(rank, total)) +
+ geom_point() +
+ geom_hline(data = line_df,
+ aes(color = cutoff,
+ yintercept = value),
+ lty = 2) +
+ scale_x_log10() +
+ scale_y_log10() +
+ labs(y = "Total UMI count")
```
 @@ -92,6 +99,26 @@ The distribution of total counts (called the unique molecular identifier or UMI
A simple approach would be to apply a threshold on the total count to only retain those barcodes with large totals. However, this may unnecessarily discard libraries derived from cell types with low RNA content.
+:::: challenge
+
+What is the median number of total counts in the raw data?
+
+::: solution
+
+
+``` r
+median(bcrank$total)
+```
+
+``` output
+[1] 2
+```
+
+Just 2! Clearly many barcodes produce practically no output.
+:::
+
+::::
+
### Testing for empty droplets
A better approach is to test whether the expression profile for each cell barcode is significantly different from the ambient RNA pool[^1]. Any significant deviation indicates that the barcode corresponds to a cell-containing droplet. This allows us to discriminate between well-sequenced empty droplets and droplets derived from cells with little RNA, both of which would have similar total counts.
@@ -182,8 +209,11 @@ library(EnsDb.Mmusculus.v79)
``` r
-chr.loc <- mapIds(EnsDb.Mmusculus.v79, keys=rownames(sce),
- keytype="GENEID", column="SEQNAME")
+chr.loc <- mapIds(EnsDb.Mmusculus.v79,
+ keys = rownames(sce),
+ keytype = "GENEID",
+ column = "SEQNAME")
+
is.mito <- which(chr.loc=="MT")
```
@@ -273,35 +303,56 @@ summary(df$subsets_Mito_percent)
0.000 1.155 1.608 5.079 2.182 66.968
```
-This can be achieved with the `perCellQCFilters` function. By default, we consider a value to be an outlier if it is more than 3 median absolute deviations (MADs) from the median in the "problematic" direction. This is loosely motivated by the fact that such a filter will retain 99% of non-outlier values that follow a normal distribution.
+We can use the `perCellQCFilters` function to apply a set of common adaptive filters to identify low-quality cells. By default, we consider a value to be an outlier if it is more than 3 median absolute deviations (MADs) from the median in the "problematic" direction. This is loosely motivated by the fact that such a filter will retain 99% of non-outlier values that follow a normal distribution.
``` r
reasons <- perCellQCFilters(df, sub.fields="subsets_Mito_percent")
-colSums(as.matrix(reasons))
+reasons
```
``` output
- low_lib_size low_n_features high_subsets_Mito_percent
- 601 654 484
- discard
- 694
+DataFrame with 3131 rows and 4 columns
+ low_lib_size low_n_features high_subsets_Mito_percent discard
+
+1 FALSE FALSE FALSE FALSE
+2 FALSE FALSE FALSE FALSE
+3 FALSE FALSE FALSE FALSE
+4 FALSE FALSE FALSE FALSE
+5 TRUE TRUE FALSE TRUE
+... ... ... ... ...
+3127 FALSE FALSE FALSE FALSE
+3128 TRUE TRUE TRUE TRUE
+3129 TRUE TRUE TRUE TRUE
+3130 TRUE TRUE FALSE TRUE
+3131 TRUE TRUE TRUE TRUE
+```
+
+``` r
+sce$discard <- reasons$discard
```
+:::: challenge
+
+We've removed empty cells and low-quality cells to be discarded. How many cells are we left with at this point?
+
+::: solution
+
``` r
-summary(reasons$discard)
+table(sce$discard)
```
``` output
- Mode FALSE TRUE
-logical 2437 694
-```
-``` r
-# include in object
-sce$discard <- reasons$discard
+FALSE TRUE
+ 2437 694
```
+There are 2437 cells that *haven't* been flagged to be discarded, so that's how many we have left.
+:::
+
+::::
+
### Diagnostic plots
It is always a good idea to check the distribution of the QC metrics and to visualize the cells that were removed, to identify possible problems with the procedure.
@@ -314,19 +365,19 @@ library(scater)
plotColData(sce, y = "sum", colour_by = "discard") + ggtitle("Total count")
```
-
@@ -92,6 +99,26 @@ The distribution of total counts (called the unique molecular identifier or UMI
A simple approach would be to apply a threshold on the total count to only retain those barcodes with large totals. However, this may unnecessarily discard libraries derived from cell types with low RNA content.
+:::: challenge
+
+What is the median number of total counts in the raw data?
+
+::: solution
+
+
+``` r
+median(bcrank$total)
+```
+
+``` output
+[1] 2
+```
+
+Just 2! Clearly many barcodes produce practically no output.
+:::
+
+::::
+
### Testing for empty droplets
A better approach is to test whether the expression profile for each cell barcode is significantly different from the ambient RNA pool[^1]. Any significant deviation indicates that the barcode corresponds to a cell-containing droplet. This allows us to discriminate between well-sequenced empty droplets and droplets derived from cells with little RNA, both of which would have similar total counts.
@@ -182,8 +209,11 @@ library(EnsDb.Mmusculus.v79)
``` r
-chr.loc <- mapIds(EnsDb.Mmusculus.v79, keys=rownames(sce),
- keytype="GENEID", column="SEQNAME")
+chr.loc <- mapIds(EnsDb.Mmusculus.v79,
+ keys = rownames(sce),
+ keytype = "GENEID",
+ column = "SEQNAME")
+
is.mito <- which(chr.loc=="MT")
```
@@ -273,35 +303,56 @@ summary(df$subsets_Mito_percent)
0.000 1.155 1.608 5.079 2.182 66.968
```
-This can be achieved with the `perCellQCFilters` function. By default, we consider a value to be an outlier if it is more than 3 median absolute deviations (MADs) from the median in the "problematic" direction. This is loosely motivated by the fact that such a filter will retain 99% of non-outlier values that follow a normal distribution.
+We can use the `perCellQCFilters` function to apply a set of common adaptive filters to identify low-quality cells. By default, we consider a value to be an outlier if it is more than 3 median absolute deviations (MADs) from the median in the "problematic" direction. This is loosely motivated by the fact that such a filter will retain 99% of non-outlier values that follow a normal distribution.
``` r
reasons <- perCellQCFilters(df, sub.fields="subsets_Mito_percent")
-colSums(as.matrix(reasons))
+reasons
```
``` output
- low_lib_size low_n_features high_subsets_Mito_percent
- 601 654 484
- discard
- 694
+DataFrame with 3131 rows and 4 columns
+ low_lib_size low_n_features high_subsets_Mito_percent discard
+
+1 FALSE FALSE FALSE FALSE
+2 FALSE FALSE FALSE FALSE
+3 FALSE FALSE FALSE FALSE
+4 FALSE FALSE FALSE FALSE
+5 TRUE TRUE FALSE TRUE
+... ... ... ... ...
+3127 FALSE FALSE FALSE FALSE
+3128 TRUE TRUE TRUE TRUE
+3129 TRUE TRUE TRUE TRUE
+3130 TRUE TRUE FALSE TRUE
+3131 TRUE TRUE TRUE TRUE
+```
+
+``` r
+sce$discard <- reasons$discard
```
+:::: challenge
+
+We've removed empty cells and low-quality cells to be discarded. How many cells are we left with at this point?
+
+::: solution
+
``` r
-summary(reasons$discard)
+table(sce$discard)
```
``` output
- Mode FALSE TRUE
-logical 2437 694
-```
-``` r
-# include in object
-sce$discard <- reasons$discard
+FALSE TRUE
+ 2437 694
```
+There are 2437 cells that *haven't* been flagged to be discarded, so that's how many we have left.
+:::
+
+::::
+
### Diagnostic plots
It is always a good idea to check the distribution of the QC metrics and to visualize the cells that were removed, to identify possible problems with the procedure.
@@ -314,19 +365,19 @@ library(scater)
plotColData(sce, y = "sum", colour_by = "discard") + ggtitle("Total count")
```
- +
+ ``` r
plotColData(sce, y = "detected", colour_by = "discard") + ggtitle("Detected features")
```
-
``` r
plotColData(sce, y = "detected", colour_by = "discard") + ggtitle("Detected features")
```
- +
+ ``` r
plotColData(sce, y = "subsets_Mito_percent", colour_by = "discard") + ggtitle("Mito percent")
```
-
``` r
plotColData(sce, y = "subsets_Mito_percent", colour_by = "discard") + ggtitle("Mito percent")
```
- +
+ While the univariate distribution of QC metrics can give some insight on the quality of the sample, often looking at the bivariate distribution of QC metrics is useful, e.g., to confirm that there are no cells with both large total counts and large mitochondrial counts, to ensure that we are not inadvertently removing high-quality cells that happen to be highly metabolically active.
@@ -335,7 +386,7 @@ While the univariate distribution of QC metrics can give some insight on the qua
plotColData(sce, x="sum", y="subsets_Mito_percent", colour_by="discard")
```
-
While the univariate distribution of QC metrics can give some insight on the quality of the sample, often looking at the bivariate distribution of QC metrics is useful, e.g., to confirm that there are no cells with both large total counts and large mitochondrial counts, to ensure that we are not inadvertently removing high-quality cells that happen to be highly metabolically active.
@@ -335,7 +386,7 @@ While the univariate distribution of QC metrics can give some insight on the qua
plotColData(sce, x="sum", y="subsets_Mito_percent", colour_by="discard")
```
- +
+ It could also be a good idea to perform a differential expression analysis between retained and discarded cells to check wether we are removing an unusual cell population rather than low-quality libraries (see [Section 1.5 of OSCA advanced](https://bioconductor.org/books/release/OSCA.advanced/quality-control-redux.html#qc-discard-cell-types)).
@@ -386,15 +437,19 @@ summary(lib.sf)
```
``` r
-hist(log10(lib.sf), xlab="Log10[Size factor]", col='grey80', breaks = 30)
+sf_df = data.frame(size_factor = lib.sf)
+
+ggplot(sf_df, aes(size_factor)) +
+ geom_histogram() +
+ scale_x_log10()
```
-
+
It could also be a good idea to perform a differential expression analysis between retained and discarded cells to check wether we are removing an unusual cell population rather than low-quality libraries (see [Section 1.5 of OSCA advanced](https://bioconductor.org/books/release/OSCA.advanced/quality-control-redux.html#qc-discard-cell-types)).
@@ -386,15 +437,19 @@ summary(lib.sf)
```
``` r
-hist(log10(lib.sf), xlab="Log10[Size factor]", col='grey80', breaks = 30)
+sf_df = data.frame(size_factor = lib.sf)
+
+ggplot(sf_df, aes(size_factor)) +
+ geom_histogram() +
+ scale_x_log10()
```
-
+ ### Normalization by deconvolution
-Library size normalization is not optimal, as it assumes that the total sum of UMI counts differ between cells only for technical and not biological reason. This can be a problem if a highly-expressed subset of genes is differentially expressed between cells or cell types.
+Library size normalization is not optimal, as it assumes that the total sum of UMI counts differ between cells only for technical and not biological reasons. This can be a problem if a highly-expressed subset of genes is differentially expressed between cells or cell types.
Several robust normalization methods have been proposed for bulk RNA-seq. However, these methods may underperform in single-cell data due to the dominance of low and zero counts.
To overcome this, one solution is to _pool_ counts from many cells to increase the size of the counts for accurate size factor estimation[^3]. Pool-based size factors are then _deconvolved_ into cell-based factors for normalization of each cell's expression profile.
@@ -429,13 +484,17 @@ summary(deconv.sf)
```
``` r
-plot(lib.sf, deconv.sf, xlab="Library size factor",
- ylab="Deconvolution size factor", log='xy', pch=16,
- col=as.integer(clust))
-abline(a=0, b=1, col="red")
+sf_df$deconv_sf = deconv.sf
+sf_df$clust = clust
+
+ggplot(sf_df, aes(size_factor, deconv_sf)) +
+ geom_abline() +
+ geom_point(aes(color = clust)) +
+ scale_x_log10() +
+ scale_y_log10()
```
-
+
### Normalization by deconvolution
-Library size normalization is not optimal, as it assumes that the total sum of UMI counts differ between cells only for technical and not biological reason. This can be a problem if a highly-expressed subset of genes is differentially expressed between cells or cell types.
+Library size normalization is not optimal, as it assumes that the total sum of UMI counts differ between cells only for technical and not biological reasons. This can be a problem if a highly-expressed subset of genes is differentially expressed between cells or cell types.
Several robust normalization methods have been proposed for bulk RNA-seq. However, these methods may underperform in single-cell data due to the dominance of low and zero counts.
To overcome this, one solution is to _pool_ counts from many cells to increase the size of the counts for accurate size factor estimation[^3]. Pool-based size factors are then _deconvolved_ into cell-based factors for normalization of each cell's expression profile.
@@ -429,13 +484,17 @@ summary(deconv.sf)
```
``` r
-plot(lib.sf, deconv.sf, xlab="Library size factor",
- ylab="Deconvolution size factor", log='xy', pch=16,
- col=as.integer(clust))
-abline(a=0, b=1, col="red")
+sf_df$deconv_sf = deconv.sf
+sf_df$clust = clust
+
+ggplot(sf_df, aes(size_factor, deconv_sf)) +
+ geom_abline() +
+ geom_point(aes(color = clust)) +
+ scale_x_log10() +
+ scale_y_log10()
```
-
+ Once we have computed the size factors, we compute the normalized expression values for each cell by dividing the count for each gene with the appropriate size factor for that cell. Since we are typically going to work with log-transformed counts, the function `logNormCounts` also log-transforms the normalized values, creating a new assay called `logcounts`.
@@ -462,6 +521,18 @@ mainExpName: NULL
altExpNames(0):
```
+:::: challenge
+
+Some sophisticated experiments perform additional steps so that they can estimate size factors from so-called "spike-ins". Judging by the name, what do you think "spike-ins" are, and what additional steps are required to use them?
+
+::: solution
+
+Spike-ins are deliberately introduced exogeneous RNA from an exotic or synthetic source at a known concentration. This provides a known signal to normalize to. Exotic or synthetic RNA (e.g. soil bacteria RNA in a study of human cells) is used in order to avoid confusing spike-in RNA with sample RNA. This has the obvious advantage of accounting for cell-wise variation, but adds additional sample-preparation work.
+
+:::
+
+::::
+
## Feature Selection
The typical next steps in the analysis of single-cell data are dimensionality reduction and clustering, which involve measuring the similarity between cells.
@@ -479,12 +550,18 @@ Calculation of the per-gene variance is simple but feature selection requires mo
dec.sce <- modelGeneVar(sce)
fit.sce <- metadata(dec.sce)
-plot(fit.sce$mean, fit.sce$var, xlab = "Mean of log-expression",
- ylab = "Variance of log-expression")
-curve(fit.sce$trend(x), col = "dodgerblue", add = TRUE, lwd = 2)
+mean_var_df = data.frame(mean = fit.sce$mean,
+ var = fit.sce$var)
+
+ggplot(mean_var_df, aes(mean, var)) +
+ geom_point() +
+ geom_function(fun = fit.sce$trend,
+ color = "dodgerblue") +
+ labs(x = "Mean of log-expression",
+ y = "Variance of log-expression")
```
-
+
Once we have computed the size factors, we compute the normalized expression values for each cell by dividing the count for each gene with the appropriate size factor for that cell. Since we are typically going to work with log-transformed counts, the function `logNormCounts` also log-transforms the normalized values, creating a new assay called `logcounts`.
@@ -462,6 +521,18 @@ mainExpName: NULL
altExpNames(0):
```
+:::: challenge
+
+Some sophisticated experiments perform additional steps so that they can estimate size factors from so-called "spike-ins". Judging by the name, what do you think "spike-ins" are, and what additional steps are required to use them?
+
+::: solution
+
+Spike-ins are deliberately introduced exogeneous RNA from an exotic or synthetic source at a known concentration. This provides a known signal to normalize to. Exotic or synthetic RNA (e.g. soil bacteria RNA in a study of human cells) is used in order to avoid confusing spike-in RNA with sample RNA. This has the obvious advantage of accounting for cell-wise variation, but adds additional sample-preparation work.
+
+:::
+
+::::
+
## Feature Selection
The typical next steps in the analysis of single-cell data are dimensionality reduction and clustering, which involve measuring the similarity between cells.
@@ -479,12 +550,18 @@ Calculation of the per-gene variance is simple but feature selection requires mo
dec.sce <- modelGeneVar(sce)
fit.sce <- metadata(dec.sce)
-plot(fit.sce$mean, fit.sce$var, xlab = "Mean of log-expression",
- ylab = "Variance of log-expression")
-curve(fit.sce$trend(x), col = "dodgerblue", add = TRUE, lwd = 2)
+mean_var_df = data.frame(mean = fit.sce$mean,
+ var = fit.sce$var)
+
+ggplot(mean_var_df, aes(mean, var)) +
+ geom_point() +
+ geom_function(fun = fit.sce$trend,
+ color = "dodgerblue") +
+ labs(x = "Mean of log-expression",
+ y = "Variance of log-expression")
```
-
+ The blue line represents the uninteresting "technical" variance for any given gene abundance. The genes with a lot of additional variance exhibit interesting "biological" variation.
@@ -503,6 +580,11 @@ head(hvg.sce.var)
[4] "ENSMUSG00000052187" "ENSMUSG00000048583" "ENSMUSG00000051855"
```
+:::: challenge
+
+Run an internet search for some of the most highly variable genes we've identified here. See if you can identify the type of protein they produce or what sort of process they're involved in. Do they make biological sense to you?
+
+::::
## Dimensionality Reduction
@@ -544,11 +626,18 @@ By default, `runPCA` computes the first 50 principal components. We can check ho
``` r
-percent.var <- attr(reducedDim(sce), "percentVar")
-plot(percent.var, log="y", xlab="PC", ylab="Variance explained (%)")
+pct_var_df = data.frame(PC = 1:50,
+ pct_var = attr(reducedDim(sce), "percentVar"))
+
+ggplot(pct_var_df,
+ aes(PC, pct_var)) +
+ geom_point() +
+ labs(y = "Variance explained (%)")
```
-
The blue line represents the uninteresting "technical" variance for any given gene abundance. The genes with a lot of additional variance exhibit interesting "biological" variation.
@@ -503,6 +580,11 @@ head(hvg.sce.var)
[4] "ENSMUSG00000052187" "ENSMUSG00000048583" "ENSMUSG00000051855"
```
+:::: challenge
+
+Run an internet search for some of the most highly variable genes we've identified here. See if you can identify the type of protein they produce or what sort of process they're involved in. Do they make biological sense to you?
+
+::::
## Dimensionality Reduction
@@ -544,11 +626,18 @@ By default, `runPCA` computes the first 50 principal components. We can check ho
``` r
-percent.var <- attr(reducedDim(sce), "percentVar")
-plot(percent.var, log="y", xlab="PC", ylab="Variance explained (%)")
+pct_var_df = data.frame(PC = 1:50,
+ pct_var = attr(reducedDim(sce), "percentVar"))
+
+ggplot(pct_var_df,
+ aes(PC, pct_var)) +
+ geom_point() +
+ labs(y = "Variance explained (%)")
```
- +
+ +
+You can see the first two PCs capture the largest amount of variation, but in this case you have to take the first 8 PCs before you've captured 50% of the total.
And we can of course visualize the first 2-3 components, perhaps color-coding each point by an interesting feature, in this case the total number of UMIs per cell.
@@ -557,19 +646,22 @@ And we can of course visualize the first 2-3 components, perhaps color-coding ea
plotPCA(sce, colour_by="sum")
```
-
+
+You can see the first two PCs capture the largest amount of variation, but in this case you have to take the first 8 PCs before you've captured 50% of the total.
And we can of course visualize the first 2-3 components, perhaps color-coding each point by an interesting feature, in this case the total number of UMIs per cell.
@@ -557,19 +646,22 @@ And we can of course visualize the first 2-3 components, perhaps color-coding ea
plotPCA(sce, colour_by="sum")
```
- +
+ +
+It can be helpful to compare pairs of PCs. This can be done with the `ncomponents` argument to `plotReducedDim()`. For example if one batch or cell type splits off on a particular PC, this can help visualize the effect of that.
+
``` r
plotReducedDim(sce, dimred="PCA", ncomponents=3)
```
-
+
+It can be helpful to compare pairs of PCs. This can be done with the `ncomponents` argument to `plotReducedDim()`. For example if one batch or cell type splits off on a particular PC, this can help visualize the effect of that.
+
``` r
plotReducedDim(sce, dimred="PCA", ncomponents=3)
```
- +
+ ### Non-linear methods
While PCA is a simple and effective way to visualize (and interpret!) scRNA-seq data, non-linear methods such as t-SNE (_t-stochastic neighbor embedding_) and UMAP (_uniform manifold approximation and projection_) have gained much popularity in the literature.
-These methods attempt to find a low-dimensional representation of the data that preserves the distances between each point and its neighbors in the high-dimensional space.
+These methods attempt to find a low-dimensional representation of the data that attempt to preserve pair-wise distance and structure in high-dimensional gene space as best as possible.
``` r
@@ -578,7 +670,7 @@ sce <- runTSNE(sce, dimred="PCA")
plotTSNE(sce)
```
-
+
### Non-linear methods
While PCA is a simple and effective way to visualize (and interpret!) scRNA-seq data, non-linear methods such as t-SNE (_t-stochastic neighbor embedding_) and UMAP (_uniform manifold approximation and projection_) have gained much popularity in the literature.
-These methods attempt to find a low-dimensional representation of the data that preserves the distances between each point and its neighbors in the high-dimensional space.
+These methods attempt to find a low-dimensional representation of the data that attempt to preserve pair-wise distance and structure in high-dimensional gene space as best as possible.
``` r
@@ -578,7 +670,7 @@ sce <- runTSNE(sce, dimred="PCA")
plotTSNE(sce)
```
-
+ ``` r
@@ -587,13 +679,13 @@ sce <- runUMAP(sce, dimred="PCA")
plotUMAP(sce)
```
-
+
``` r
@@ -587,13 +679,13 @@ sce <- runUMAP(sce, dimred="PCA")
plotUMAP(sce)
```
-
+ It is easy to over-interpret t-SNE and UMAP plots. We note that the relative sizes and positions of the visual clusters may be misleading, as they tend to inflate dense clusters and compress sparse ones, such that we cannot use the size as a measure of subpopulation heterogeneity.
In addition, these methods are not guaranteed to preserve the global structure of the data (e.g., the relative locations of non-neighboring clusters), such that we cannot use their positions to determine relationships between distant clusters.
-Note that the `sce` object now includes all the computed dimensionality reduced representations of the data for ease of reusing and replotting without the need for recomputing.
+Note that the `sce` object now includes all the computed dimensionality reduced representations of the data for ease of reusing and replotting without the need for recomputing. Note the added `reducedDimNames` row when printing `sce` here:
``` r
@@ -616,9 +708,19 @@ mainExpName: NULL
altExpNames(0):
```
-Despite their shortcomings, t-SNE and UMAP may be useful visualization techniques.
+Despite their shortcomings, t-SNE and UMAP can be useful visualization techniques.
When using them, it is important to consider that they are stochastic methods that involve a random component (each run will lead to different plots) and that there are key parameters to be set that change the results substantially (e.g., the "perplexity" parameter of t-SNE).
+:::: challenge
+
+Can dimensionality reduction techniques provide a perfectly accurate representation of the data?
+
+::: solution
+Mathematically, this would require the data to fall on a two-dimensional plane (for linear methods like PCA) or a smooth 2D manifold (for methods like UMAP). You can be confident that this will never happen in real-world data, so the reduction from ~2500-dimensional gene space to two-dimensional plot space always involves some degree of information loss.
+:::
+
+::::
+
## Doublet identification
_Doublets_ are artifactual libraries generated from two cells. They typically arise due to errors in cell sorting or capture. Specifically, in droplet-based protocols, it may happen that two cells are captured in the same droplet.
@@ -629,7 +731,7 @@ It is not easy to computationally identify doublets as they can be hard to disti
There are several computational methods to identify doublets; we describe only one here based on in-silico simulation of doublets.
-### Computing doublet desities
+### Computing doublet densities
At a high level, the algorithm can be defined by the following steps:
@@ -663,7 +765,7 @@ sce$DoubletScore <- dbl.dens
plotTSNE(sce, colour_by="DoubletScore")
```
-
+
It is easy to over-interpret t-SNE and UMAP plots. We note that the relative sizes and positions of the visual clusters may be misleading, as they tend to inflate dense clusters and compress sparse ones, such that we cannot use the size as a measure of subpopulation heterogeneity.
In addition, these methods are not guaranteed to preserve the global structure of the data (e.g., the relative locations of non-neighboring clusters), such that we cannot use their positions to determine relationships between distant clusters.
-Note that the `sce` object now includes all the computed dimensionality reduced representations of the data for ease of reusing and replotting without the need for recomputing.
+Note that the `sce` object now includes all the computed dimensionality reduced representations of the data for ease of reusing and replotting without the need for recomputing. Note the added `reducedDimNames` row when printing `sce` here:
``` r
@@ -616,9 +708,19 @@ mainExpName: NULL
altExpNames(0):
```
-Despite their shortcomings, t-SNE and UMAP may be useful visualization techniques.
+Despite their shortcomings, t-SNE and UMAP can be useful visualization techniques.
When using them, it is important to consider that they are stochastic methods that involve a random component (each run will lead to different plots) and that there are key parameters to be set that change the results substantially (e.g., the "perplexity" parameter of t-SNE).
+:::: challenge
+
+Can dimensionality reduction techniques provide a perfectly accurate representation of the data?
+
+::: solution
+Mathematically, this would require the data to fall on a two-dimensional plane (for linear methods like PCA) or a smooth 2D manifold (for methods like UMAP). You can be confident that this will never happen in real-world data, so the reduction from ~2500-dimensional gene space to two-dimensional plot space always involves some degree of information loss.
+:::
+
+::::
+
## Doublet identification
_Doublets_ are artifactual libraries generated from two cells. They typically arise due to errors in cell sorting or capture. Specifically, in droplet-based protocols, it may happen that two cells are captured in the same droplet.
@@ -629,7 +731,7 @@ It is not easy to computationally identify doublets as they can be hard to disti
There are several computational methods to identify doublets; we describe only one here based on in-silico simulation of doublets.
-### Computing doublet desities
+### Computing doublet densities
At a high level, the algorithm can be defined by the following steps:
@@ -663,7 +765,7 @@ sce$DoubletScore <- dbl.dens
plotTSNE(sce, colour_by="DoubletScore")
```
-
+ We can explicitly convert this into doublet calls by identifying large outliers for the score within each sample. Here we use the "griffiths" method to do so.
@@ -684,13 +786,13 @@ sce$doublet <- dbl.calls
plotColData(sce, y="DoubletScore", colour_by="doublet")
```
-
+
We can explicitly convert this into doublet calls by identifying large outliers for the score within each sample. Here we use the "griffiths" method to do so.
@@ -684,13 +786,13 @@ sce$doublet <- dbl.calls
plotColData(sce, y="DoubletScore", colour_by="doublet")
```
-
+ ``` r
plotTSNE(sce, colour_by="doublet")
```
-
``` r
plotTSNE(sce, colour_by="doublet")
```
- +
+ One way to determine whether a cell is in a real transient state or it is a doublet is to check the number of detected genes and total UMI counts.
@@ -699,13 +801,13 @@ One way to determine whether a cell is in a real transient state or it is a doub
plotColData(sce, "detected", "sum", colour_by = "DoubletScore")
```
-
One way to determine whether a cell is in a real transient state or it is a doublet is to check the number of detected genes and total UMI counts.
@@ -699,13 +801,13 @@ One way to determine whether a cell is in a real transient state or it is a doub
plotColData(sce, "detected", "sum", colour_by = "DoubletScore")
```
- +
+ ``` r
plotColData(sce, "detected", "sum", colour_by = "doublet")
```
-
``` r
plotColData(sce, "detected", "sum", colour_by = "doublet")
```
- +
+ In this case, we only have a few doublets at the periphery of some clusters. It could be fine to keep the doublets in the analysis, but it may be safer to discard them to avoid biases in downstream analysis (e.g., differential expression).
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-1.png b/fig/eda_qc-rendered-unnamed-chunk-12-1.png
new file mode 100644
index 0000000..b671ad6
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-2.png b/fig/eda_qc-rendered-unnamed-chunk-12-2.png
new file mode 100644
index 0000000..3a10b39
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-2.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-3.png b/fig/eda_qc-rendered-unnamed-chunk-12-3.png
new file mode 100644
index 0000000..bd6f9e5
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-3.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-13-1.png b/fig/eda_qc-rendered-unnamed-chunk-13-1.png
index 702775d..c6a1259 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-13-1.png and b/fig/eda_qc-rendered-unnamed-chunk-13-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-15-1.png b/fig/eda_qc-rendered-unnamed-chunk-15-1.png
index 3170cac..6d1ccbc 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-15-1.png and b/fig/eda_qc-rendered-unnamed-chunk-15-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-17-1.png b/fig/eda_qc-rendered-unnamed-chunk-17-1.png
index 78944cf..0893920 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-17-1.png and b/fig/eda_qc-rendered-unnamed-chunk-17-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-19-1.png b/fig/eda_qc-rendered-unnamed-chunk-19-1.png
new file mode 100644
index 0000000..181c979
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-19-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-2-1.png b/fig/eda_qc-rendered-unnamed-chunk-2-1.png
index 2cd2c3c..9e8cf49 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-2-1.png and b/fig/eda_qc-rendered-unnamed-chunk-2-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-22-1.png b/fig/eda_qc-rendered-unnamed-chunk-22-1.png
index e3fb09e..a8555d0 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-22-1.png and b/fig/eda_qc-rendered-unnamed-chunk-22-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-23-1.png b/fig/eda_qc-rendered-unnamed-chunk-23-1.png
index ff232ae..de7cec1 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-23-1.png and b/fig/eda_qc-rendered-unnamed-chunk-23-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-24-1.png b/fig/eda_qc-rendered-unnamed-chunk-24-1.png
new file mode 100644
index 0000000..c803807
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-24-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-25-1.png b/fig/eda_qc-rendered-unnamed-chunk-25-1.png
index 433225a..e3fb09e 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-25-1.png and b/fig/eda_qc-rendered-unnamed-chunk-25-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-26-1.png b/fig/eda_qc-rendered-unnamed-chunk-26-1.png
index 8cf9120..ff232ae 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-26-1.png and b/fig/eda_qc-rendered-unnamed-chunk-26-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-28-1.png b/fig/eda_qc-rendered-unnamed-chunk-28-1.png
new file mode 100644
index 0000000..433225a
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-28-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-29-1.png b/fig/eda_qc-rendered-unnamed-chunk-29-1.png
new file mode 100644
index 0000000..8cf9120
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-29-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-29-2.png b/fig/eda_qc-rendered-unnamed-chunk-29-2.png
new file mode 100644
index 0000000..2ab61c9
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-29-2.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-30-1.png b/fig/eda_qc-rendered-unnamed-chunk-30-1.png
new file mode 100644
index 0000000..f9d0b4d
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-30-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-30-2.png b/fig/eda_qc-rendered-unnamed-chunk-30-2.png
new file mode 100644
index 0000000..9cc8b17
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-30-2.png differ
diff --git a/fig/intro-sce-rendered-unnamed-chunk-13-1.png b/fig/intro-sce-rendered-unnamed-chunk-13-1.png
new file mode 100644
index 0000000..cd3f489
Binary files /dev/null and b/fig/intro-sce-rendered-unnamed-chunk-13-1.png differ
diff --git a/intro-sce.md b/intro-sce.md
index bb62274..6380cde 100644
--- a/intro-sce.md
+++ b/intro-sce.md
@@ -30,15 +30,15 @@ library(SingleCellExperiment)
library(MouseGastrulationData)
```
+It's normal to see lot of startup messages when loading these packages.
+
## Bioconductor
### Overview
Within the R ecosystem, the Bioconductor project provides tools for the analysis and comprehension of high-throughput genomics data.
The scope of the project covers microarray data, various forms of sequencing (RNA-seq, ChIP-seq, bisulfite, genotyping, etc.), proteomics, flow cytometry and more.
-One of Bioconductor's main selling points is the use of common data structures to promote interoperability between packages,
-allowing code written by different people (from different organizations, in different countries) to work together seamlessly in complex analyses.
-By extending R to genomics, Bioconductor serves as a powerful addition to the computational biologist's toolkit.
+One of Bioconductor's main selling points is the use of common data structures to promote interoperability between packages, allowing code written by different people (from different organizations, in different countries) to work together seamlessly in complex analyses.
### Installing Bioconductor Packages
@@ -63,9 +63,6 @@ For example, the code chunk below uses this approach to install the *[SingleCell
``` r
-## The command below is a one-line shortcut for:
-## library(BiocManager)
-## install("SingleCellExperiment")
BiocManager::install("SingleCellExperiment")
```
@@ -102,13 +99,15 @@ This will check for more recent versions of each package (within a Bioconductor
BiocManager::install()
```
+Be careful: if you have a lot of packages to update, this can take a long time.
+
## The `SingleCellExperiment` class
One of the main strengths of the Bioconductor project lies in the use of a common data infrastructure that powers interoperability across packages.
Users should be able to analyze their data using functions from different Bioconductor packages without the need to convert between formats. To this end, the `SingleCellExperiment` class (from the _SingleCellExperiment_ package) serves as the common currency for data exchange across 70+ single-cell-related Bioconductor packages.
-This class implements a data structure that stores all aspects of our single-cell data - gene-by-cell expression data, per-cell metadata and per-gene annotation - and manipulate them in a synchronized manner.
+This class implements a data structure that stores all aspects of our single-cell data - gene-by-cell expression data, cell-wise metadata, and gene-wise annotation - and lets us manipulate them in an organized manner.
In this case, we only have a few doublets at the periphery of some clusters. It could be fine to keep the doublets in the analysis, but it may be safer to discard them to avoid biases in downstream analysis (e.g., differential expression).
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-1.png b/fig/eda_qc-rendered-unnamed-chunk-12-1.png
new file mode 100644
index 0000000..b671ad6
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-2.png b/fig/eda_qc-rendered-unnamed-chunk-12-2.png
new file mode 100644
index 0000000..3a10b39
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-2.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-12-3.png b/fig/eda_qc-rendered-unnamed-chunk-12-3.png
new file mode 100644
index 0000000..bd6f9e5
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-12-3.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-13-1.png b/fig/eda_qc-rendered-unnamed-chunk-13-1.png
index 702775d..c6a1259 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-13-1.png and b/fig/eda_qc-rendered-unnamed-chunk-13-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-15-1.png b/fig/eda_qc-rendered-unnamed-chunk-15-1.png
index 3170cac..6d1ccbc 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-15-1.png and b/fig/eda_qc-rendered-unnamed-chunk-15-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-17-1.png b/fig/eda_qc-rendered-unnamed-chunk-17-1.png
index 78944cf..0893920 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-17-1.png and b/fig/eda_qc-rendered-unnamed-chunk-17-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-19-1.png b/fig/eda_qc-rendered-unnamed-chunk-19-1.png
new file mode 100644
index 0000000..181c979
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-19-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-2-1.png b/fig/eda_qc-rendered-unnamed-chunk-2-1.png
index 2cd2c3c..9e8cf49 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-2-1.png and b/fig/eda_qc-rendered-unnamed-chunk-2-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-22-1.png b/fig/eda_qc-rendered-unnamed-chunk-22-1.png
index e3fb09e..a8555d0 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-22-1.png and b/fig/eda_qc-rendered-unnamed-chunk-22-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-23-1.png b/fig/eda_qc-rendered-unnamed-chunk-23-1.png
index ff232ae..de7cec1 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-23-1.png and b/fig/eda_qc-rendered-unnamed-chunk-23-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-24-1.png b/fig/eda_qc-rendered-unnamed-chunk-24-1.png
new file mode 100644
index 0000000..c803807
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-24-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-25-1.png b/fig/eda_qc-rendered-unnamed-chunk-25-1.png
index 433225a..e3fb09e 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-25-1.png and b/fig/eda_qc-rendered-unnamed-chunk-25-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-26-1.png b/fig/eda_qc-rendered-unnamed-chunk-26-1.png
index 8cf9120..ff232ae 100644
Binary files a/fig/eda_qc-rendered-unnamed-chunk-26-1.png and b/fig/eda_qc-rendered-unnamed-chunk-26-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-28-1.png b/fig/eda_qc-rendered-unnamed-chunk-28-1.png
new file mode 100644
index 0000000..433225a
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-28-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-29-1.png b/fig/eda_qc-rendered-unnamed-chunk-29-1.png
new file mode 100644
index 0000000..8cf9120
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-29-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-29-2.png b/fig/eda_qc-rendered-unnamed-chunk-29-2.png
new file mode 100644
index 0000000..2ab61c9
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-29-2.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-30-1.png b/fig/eda_qc-rendered-unnamed-chunk-30-1.png
new file mode 100644
index 0000000..f9d0b4d
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-30-1.png differ
diff --git a/fig/eda_qc-rendered-unnamed-chunk-30-2.png b/fig/eda_qc-rendered-unnamed-chunk-30-2.png
new file mode 100644
index 0000000..9cc8b17
Binary files /dev/null and b/fig/eda_qc-rendered-unnamed-chunk-30-2.png differ
diff --git a/fig/intro-sce-rendered-unnamed-chunk-13-1.png b/fig/intro-sce-rendered-unnamed-chunk-13-1.png
new file mode 100644
index 0000000..cd3f489
Binary files /dev/null and b/fig/intro-sce-rendered-unnamed-chunk-13-1.png differ
diff --git a/intro-sce.md b/intro-sce.md
index bb62274..6380cde 100644
--- a/intro-sce.md
+++ b/intro-sce.md
@@ -30,15 +30,15 @@ library(SingleCellExperiment)
library(MouseGastrulationData)
```
+It's normal to see lot of startup messages when loading these packages.
+
## Bioconductor
### Overview
Within the R ecosystem, the Bioconductor project provides tools for the analysis and comprehension of high-throughput genomics data.
The scope of the project covers microarray data, various forms of sequencing (RNA-seq, ChIP-seq, bisulfite, genotyping, etc.), proteomics, flow cytometry and more.
-One of Bioconductor's main selling points is the use of common data structures to promote interoperability between packages,
-allowing code written by different people (from different organizations, in different countries) to work together seamlessly in complex analyses.
-By extending R to genomics, Bioconductor serves as a powerful addition to the computational biologist's toolkit.
+One of Bioconductor's main selling points is the use of common data structures to promote interoperability between packages, allowing code written by different people (from different organizations, in different countries) to work together seamlessly in complex analyses.
### Installing Bioconductor Packages
@@ -63,9 +63,6 @@ For example, the code chunk below uses this approach to install the *[SingleCell
``` r
-## The command below is a one-line shortcut for:
-## library(BiocManager)
-## install("SingleCellExperiment")
BiocManager::install("SingleCellExperiment")
```
@@ -102,13 +99,15 @@ This will check for more recent versions of each package (within a Bioconductor
BiocManager::install()
```
+Be careful: if you have a lot of packages to update, this can take a long time.
+
## The `SingleCellExperiment` class
One of the main strengths of the Bioconductor project lies in the use of a common data infrastructure that powers interoperability across packages.
Users should be able to analyze their data using functions from different Bioconductor packages without the need to convert between formats. To this end, the `SingleCellExperiment` class (from the _SingleCellExperiment_ package) serves as the common currency for data exchange across 70+ single-cell-related Bioconductor packages.
-This class implements a data structure that stores all aspects of our single-cell data - gene-by-cell expression data, per-cell metadata and per-gene annotation - and manipulate them in a synchronized manner.
+This class implements a data structure that stores all aspects of our single-cell data - gene-by-cell expression data, cell-wise metadata, and gene-wise annotation - and lets us manipulate them in an organized manner.
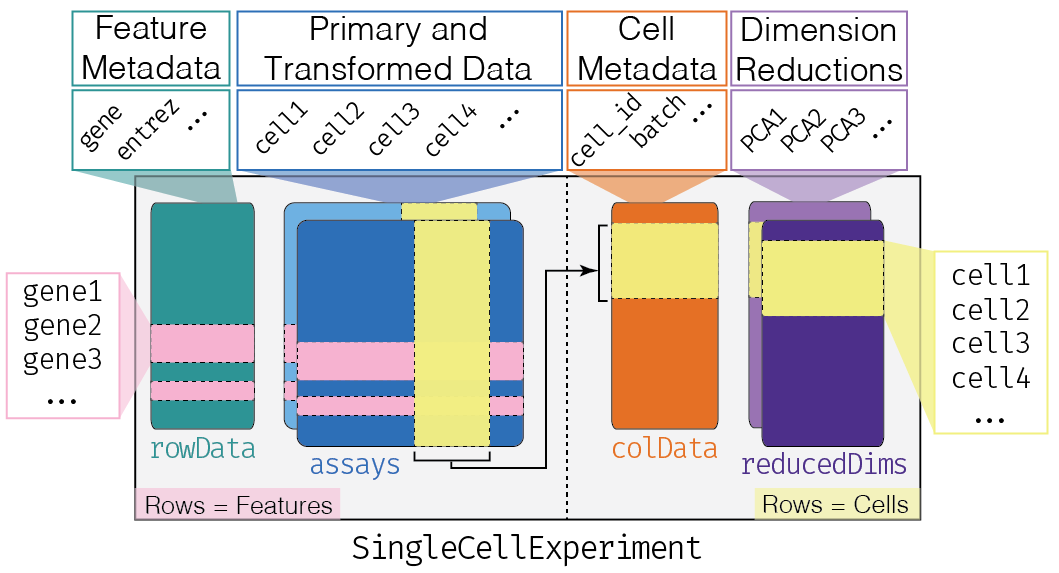 @@ -135,19 +134,18 @@ mainExpName: NULL
altExpNames(0):
```
-We can think of this (and other) class as a _container_, that contains several different pieces of data in so-called _slots_.
+We can think of this (and other) class as a _container_, that contains several different pieces of data in so-called _slots_. SingleCellExperiment objects come with dedicated methods for _getting_ and _setting_ the data in their slots.
-The _getter_ methods are used to extract information from the slots and the _setter_ methods are used to add information into the slots. These are the only ways to interact with the objects (rather than directly accessing the slots).
+Depending on the object, slots can contain different types of data (e.g., numeric matrices, lists, etc.). Here we'll review the main slots of the SingleCellExperiment class as well as their getter/setter methods.
-Depending on the object, slots can contain different types of data (e.g., numeric matrices, lists, etc.). We will here review the main slots of the SingleCellExperiment class as well as their getter/setter methods.
:::: challenge
-Before SingleCellExperiments, coders working with single cell data would sometimes keep all of these components in separate objects e.g. a matrix of counts, a data.frame of sample metadata, a data.frame of gene annotations and so on. What are the main disadvantages of this sort of "from scratch" approach?
+Before SingleCellExperiments, coders working with single cell data would sometimes keep all of these components in separate objects e.g. a matrix of counts, a data.frame of sample metadata, a data.frame of gene annotations and so on. What are the main disadvantage of this sort of "from scratch" approach?
::: solution
-1. You have to do tons of manual book-keeping! If you perform a QC step that removes dead cells, now you also have to remember to remove that same set of cells from the cell-wise metadata. Dropped un-expressed genes? Don't forget to filter the gene metadata table too.
+1. You have to do tons of book-keeping! If you perform a QC step that removes dead cells, now you also have to remember to remove that same set of cells from the cell-wise metadata. Dropped un-expressed genes? Don't forget to filter the gene metadata table too.
2. All the downstream steps have to be "from scratch" as well! If your tables have some slight format difference from those of your lab-mate, suddenly the plotting code you're trying to re-use doesn't work! Agh!
@@ -155,17 +153,17 @@ Before SingleCellExperiments, coders working with single cell data would sometim
::::
-### The `assays`
+### `assays`
This is arguably the most fundamental part of the object that contains the count matrix, and potentially other matrices with transformed data. We can access the _list_ of matrices with the `assays` function and individual matrices with the `assay` function. If one of these matrices is called "counts", we can use the special `counts` getter (and the analogous `logcounts`).
``` r
-identical(assay(sce), counts(sce))
+names(assays(sce))
```
``` output
-[1] TRUE
+[1] "counts"
```
``` r
@@ -182,7 +180,7 @@ ENSMUSG00000102343 . . .
You will notice that in this case we have a sparse matrix of class "dgTMatrix" inside the object. More generally, any "matrix-like" object can be used, e.g., dense matrices or HDF5-backed matrices (see "Working with large data").
-### The `colData` and `rowData`
+### `colData` and `rowData`
Conceptually, these are two data frames that annotate the columns and the rows of your assay, respectively.
@@ -190,131 +188,74 @@ One can interact with them as usual, e.g., by extracting columns or adding addit
``` r
-colData(sce)
+colData(sce)[1:3, 1:4]
```
``` output
-DataFrame with 2411 rows and 11 columns
- cell barcode sample stage tomato
-
-cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
-cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
-cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
-cell_9772 cell_9772 AAACCTGCATACTCTT 5 E8.5 TRUE
-cell_9773 cell_9773 AAACGGGTCAACACCA 5 E8.5 TRUE
-... ... ... ... ... ...
-cell_12175 cell_12175 TTTGGTTAGTCCGTAT 5 E8.5 TRUE
-cell_12176 cell_12176 TTTGGTTAGTGTTGAA 5 E8.5 TRUE
-cell_12177 cell_12177 TTTGGTTGTTAAAGAC 5 E8.5 TRUE
-cell_12178 cell_12178 TTTGGTTTCAGTCAGT 5 E8.5 TRUE
-cell_12179 cell_12179 TTTGGTTTCGCCATAA 5 E8.5 TRUE
- pool stage.mapped celltype.mapped closest.cell
-
-cell_9769 3 E8.25 Mesenchyme cell_24159
-cell_9770 3 E8.5 Endothelium cell_96660
-cell_9771 3 E8.5 Allantois cell_134982
-cell_9772 3 E8.5 Erythroid3 cell_133892
-cell_9773 3 E8.25 Erythroid1 cell_76296
-... ... ... ... ...
-cell_12175 3 E8.5 Erythroid3 cell_138060
-cell_12176 3 E8.5 Forebrain/Midbrain/H.. cell_72709
-cell_12177 3 E8.25 Surface ectoderm cell_100275
-cell_12178 3 E8.25 Erythroid2 cell_70906
-cell_12179 3 E8.5 Spinal cord cell_102334
- doub.density sizeFactor
-
-cell_9769 0.02985045 1.41243
-cell_9770 0.00172753 1.22757
-cell_9771 0.01338013 1.15439
-cell_9772 0.00218402 1.28676
-cell_9773 0.00211723 1.78719
-... ... ...
-cell_12175 0.00129403 1.219506
-cell_12176 0.01833074 1.095753
-cell_12177 0.03104037 0.910728
-cell_12178 0.00169483 2.061701
-cell_12179 0.03767894 1.798687
+DataFrame with 3 rows and 4 columns
+ cell barcode sample stage
+
+cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5
+cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5
+cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5
```
``` r
-rowData(sce)
+rowData(sce)[1:3, 1:2]
```
``` output
-DataFrame with 29453 rows and 2 columns
- ENSEMBL SYMBOL
-
-ENSMUSG00000051951 ENSMUSG00000051951 Xkr4
-ENSMUSG00000089699 ENSMUSG00000089699 Gm1992
-ENSMUSG00000102343 ENSMUSG00000102343 Gm37381
-ENSMUSG00000025900 ENSMUSG00000025900 Rp1
-ENSMUSG00000025902 ENSMUSG00000025902 Sox17
-... ... ...
-ENSMUSG00000095041 ENSMUSG00000095041 AC149090.1
-ENSMUSG00000063897 ENSMUSG00000063897 DHRSX
-ENSMUSG00000096730 ENSMUSG00000096730 Vmn2r122
-ENSMUSG00000095742 ENSMUSG00000095742 CAAA01147332.1
-tomato-td tomato-td tomato-td
+DataFrame with 3 rows and 2 columns
+ ENSEMBL SYMBOL
+
+ENSMUSG00000051951 ENSMUSG00000051951 Xkr4
+ENSMUSG00000089699 ENSMUSG00000089699 Gm1992
+ENSMUSG00000102343 ENSMUSG00000102343 Gm37381
```
-Note the `$` short cut.
+You can access columns of the colData with the `$` accessor to quickly add cell-wise metadata to the colData
``` r
-identical(colData(sce)$sum, sce$sum)
+sce$my_sum <- colSums(counts(sce))
+colData(sce)[1:3,]
```
``` output
-[1] TRUE
+DataFrame with 3 rows and 12 columns
+ cell barcode sample stage tomato
+
+cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
+cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
+cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
+ pool stage.mapped celltype.mapped closest.cell doub.density
+
+cell_9769 3 E8.25 Mesenchyme cell_24159 0.02985045
+cell_9770 3 E8.5 Endothelium cell_96660 0.00172753
+cell_9771 3 E8.5 Allantois cell_134982 0.01338013
+ sizeFactor my_sum
+
+cell_9769 1.41243 27577
+cell_9770 1.22757 29309
+cell_9771 1.15439 28795
```
+:::: challenge
+
+Try to add a column of gene-wise metadata to the rowData.
+
+::: solution
+
+Here we add a column called "conservation" that is just an integer sequence from 1 to the number of genes.
+
+
``` r
-sce$my_sum <- colSums(counts(sce))
-colData(sce)
+rowData(sce)$conservation = 1:nrow(sce)
```
-``` output
-DataFrame with 2411 rows and 12 columns
- cell barcode sample stage tomato
-
-cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
-cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
-cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
-cell_9772 cell_9772 AAACCTGCATACTCTT 5 E8.5 TRUE
-cell_9773 cell_9773 AAACGGGTCAACACCA 5 E8.5 TRUE
-... ... ... ... ... ...
-cell_12175 cell_12175 TTTGGTTAGTCCGTAT 5 E8.5 TRUE
-cell_12176 cell_12176 TTTGGTTAGTGTTGAA 5 E8.5 TRUE
-cell_12177 cell_12177 TTTGGTTGTTAAAGAC 5 E8.5 TRUE
-cell_12178 cell_12178 TTTGGTTTCAGTCAGT 5 E8.5 TRUE
-cell_12179 cell_12179 TTTGGTTTCGCCATAA 5 E8.5 TRUE
- pool stage.mapped celltype.mapped closest.cell
-
-cell_9769 3 E8.25 Mesenchyme cell_24159
-cell_9770 3 E8.5 Endothelium cell_96660
-cell_9771 3 E8.5 Allantois cell_134982
-cell_9772 3 E8.5 Erythroid3 cell_133892
-cell_9773 3 E8.25 Erythroid1 cell_76296
-... ... ... ... ...
-cell_12175 3 E8.5 Erythroid3 cell_138060
-cell_12176 3 E8.5 Forebrain/Midbrain/H.. cell_72709
-cell_12177 3 E8.25 Surface ectoderm cell_100275
-cell_12178 3 E8.25 Erythroid2 cell_70906
-cell_12179 3 E8.5 Spinal cord cell_102334
- doub.density sizeFactor my_sum
-
-cell_9769 0.02985045 1.41243 27577
-cell_9770 0.00172753 1.22757 29309
-cell_9771 0.01338013 1.15439 28795
-cell_9772 0.00218402 1.28676 34794
-cell_9773 0.00211723 1.78719 38300
-... ... ... ...
-cell_12175 0.00129403 1.219506 26680
-cell_12176 0.01833074 1.095753 19013
-cell_12177 0.03104037 0.910728 24627
-cell_12178 0.00169483 2.061701 46162
-cell_12179 0.03767894 1.798687 38398
-```
+:::
+
+::::
### The `reducedDims`
@@ -344,7 +285,7 @@ library(scater)
plotReducedDim(sce, "pca.corrected.E8.5", colour_by = "celltype.mapped")
```
-
@@ -135,19 +134,18 @@ mainExpName: NULL
altExpNames(0):
```
-We can think of this (and other) class as a _container_, that contains several different pieces of data in so-called _slots_.
+We can think of this (and other) class as a _container_, that contains several different pieces of data in so-called _slots_. SingleCellExperiment objects come with dedicated methods for _getting_ and _setting_ the data in their slots.
-The _getter_ methods are used to extract information from the slots and the _setter_ methods are used to add information into the slots. These are the only ways to interact with the objects (rather than directly accessing the slots).
+Depending on the object, slots can contain different types of data (e.g., numeric matrices, lists, etc.). Here we'll review the main slots of the SingleCellExperiment class as well as their getter/setter methods.
-Depending on the object, slots can contain different types of data (e.g., numeric matrices, lists, etc.). We will here review the main slots of the SingleCellExperiment class as well as their getter/setter methods.
:::: challenge
-Before SingleCellExperiments, coders working with single cell data would sometimes keep all of these components in separate objects e.g. a matrix of counts, a data.frame of sample metadata, a data.frame of gene annotations and so on. What are the main disadvantages of this sort of "from scratch" approach?
+Before SingleCellExperiments, coders working with single cell data would sometimes keep all of these components in separate objects e.g. a matrix of counts, a data.frame of sample metadata, a data.frame of gene annotations and so on. What are the main disadvantage of this sort of "from scratch" approach?
::: solution
-1. You have to do tons of manual book-keeping! If you perform a QC step that removes dead cells, now you also have to remember to remove that same set of cells from the cell-wise metadata. Dropped un-expressed genes? Don't forget to filter the gene metadata table too.
+1. You have to do tons of book-keeping! If you perform a QC step that removes dead cells, now you also have to remember to remove that same set of cells from the cell-wise metadata. Dropped un-expressed genes? Don't forget to filter the gene metadata table too.
2. All the downstream steps have to be "from scratch" as well! If your tables have some slight format difference from those of your lab-mate, suddenly the plotting code you're trying to re-use doesn't work! Agh!
@@ -155,17 +153,17 @@ Before SingleCellExperiments, coders working with single cell data would sometim
::::
-### The `assays`
+### `assays`
This is arguably the most fundamental part of the object that contains the count matrix, and potentially other matrices with transformed data. We can access the _list_ of matrices with the `assays` function and individual matrices with the `assay` function. If one of these matrices is called "counts", we can use the special `counts` getter (and the analogous `logcounts`).
``` r
-identical(assay(sce), counts(sce))
+names(assays(sce))
```
``` output
-[1] TRUE
+[1] "counts"
```
``` r
@@ -182,7 +180,7 @@ ENSMUSG00000102343 . . .
You will notice that in this case we have a sparse matrix of class "dgTMatrix" inside the object. More generally, any "matrix-like" object can be used, e.g., dense matrices or HDF5-backed matrices (see "Working with large data").
-### The `colData` and `rowData`
+### `colData` and `rowData`
Conceptually, these are two data frames that annotate the columns and the rows of your assay, respectively.
@@ -190,131 +188,74 @@ One can interact with them as usual, e.g., by extracting columns or adding addit
``` r
-colData(sce)
+colData(sce)[1:3, 1:4]
```
``` output
-DataFrame with 2411 rows and 11 columns
- cell barcode sample stage tomato
-
-cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
-cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
-cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
-cell_9772 cell_9772 AAACCTGCATACTCTT 5 E8.5 TRUE
-cell_9773 cell_9773 AAACGGGTCAACACCA 5 E8.5 TRUE
-... ... ... ... ... ...
-cell_12175 cell_12175 TTTGGTTAGTCCGTAT 5 E8.5 TRUE
-cell_12176 cell_12176 TTTGGTTAGTGTTGAA 5 E8.5 TRUE
-cell_12177 cell_12177 TTTGGTTGTTAAAGAC 5 E8.5 TRUE
-cell_12178 cell_12178 TTTGGTTTCAGTCAGT 5 E8.5 TRUE
-cell_12179 cell_12179 TTTGGTTTCGCCATAA 5 E8.5 TRUE
- pool stage.mapped celltype.mapped closest.cell
-
-cell_9769 3 E8.25 Mesenchyme cell_24159
-cell_9770 3 E8.5 Endothelium cell_96660
-cell_9771 3 E8.5 Allantois cell_134982
-cell_9772 3 E8.5 Erythroid3 cell_133892
-cell_9773 3 E8.25 Erythroid1 cell_76296
-... ... ... ... ...
-cell_12175 3 E8.5 Erythroid3 cell_138060
-cell_12176 3 E8.5 Forebrain/Midbrain/H.. cell_72709
-cell_12177 3 E8.25 Surface ectoderm cell_100275
-cell_12178 3 E8.25 Erythroid2 cell_70906
-cell_12179 3 E8.5 Spinal cord cell_102334
- doub.density sizeFactor
-
-cell_9769 0.02985045 1.41243
-cell_9770 0.00172753 1.22757
-cell_9771 0.01338013 1.15439
-cell_9772 0.00218402 1.28676
-cell_9773 0.00211723 1.78719
-... ... ...
-cell_12175 0.00129403 1.219506
-cell_12176 0.01833074 1.095753
-cell_12177 0.03104037 0.910728
-cell_12178 0.00169483 2.061701
-cell_12179 0.03767894 1.798687
+DataFrame with 3 rows and 4 columns
+ cell barcode sample stage
+
+cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5
+cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5
+cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5
```
``` r
-rowData(sce)
+rowData(sce)[1:3, 1:2]
```
``` output
-DataFrame with 29453 rows and 2 columns
- ENSEMBL SYMBOL
-
-ENSMUSG00000051951 ENSMUSG00000051951 Xkr4
-ENSMUSG00000089699 ENSMUSG00000089699 Gm1992
-ENSMUSG00000102343 ENSMUSG00000102343 Gm37381
-ENSMUSG00000025900 ENSMUSG00000025900 Rp1
-ENSMUSG00000025902 ENSMUSG00000025902 Sox17
-... ... ...
-ENSMUSG00000095041 ENSMUSG00000095041 AC149090.1
-ENSMUSG00000063897 ENSMUSG00000063897 DHRSX
-ENSMUSG00000096730 ENSMUSG00000096730 Vmn2r122
-ENSMUSG00000095742 ENSMUSG00000095742 CAAA01147332.1
-tomato-td tomato-td tomato-td
+DataFrame with 3 rows and 2 columns
+ ENSEMBL SYMBOL
+
+ENSMUSG00000051951 ENSMUSG00000051951 Xkr4
+ENSMUSG00000089699 ENSMUSG00000089699 Gm1992
+ENSMUSG00000102343 ENSMUSG00000102343 Gm37381
```
-Note the `$` short cut.
+You can access columns of the colData with the `$` accessor to quickly add cell-wise metadata to the colData
``` r
-identical(colData(sce)$sum, sce$sum)
+sce$my_sum <- colSums(counts(sce))
+colData(sce)[1:3,]
```
``` output
-[1] TRUE
+DataFrame with 3 rows and 12 columns
+ cell barcode sample stage tomato
+
+cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
+cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
+cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
+ pool stage.mapped celltype.mapped closest.cell doub.density
+
+cell_9769 3 E8.25 Mesenchyme cell_24159 0.02985045
+cell_9770 3 E8.5 Endothelium cell_96660 0.00172753
+cell_9771 3 E8.5 Allantois cell_134982 0.01338013
+ sizeFactor my_sum
+
+cell_9769 1.41243 27577
+cell_9770 1.22757 29309
+cell_9771 1.15439 28795
```
+:::: challenge
+
+Try to add a column of gene-wise metadata to the rowData.
+
+::: solution
+
+Here we add a column called "conservation" that is just an integer sequence from 1 to the number of genes.
+
+
``` r
-sce$my_sum <- colSums(counts(sce))
-colData(sce)
+rowData(sce)$conservation = 1:nrow(sce)
```
-``` output
-DataFrame with 2411 rows and 12 columns
- cell barcode sample stage tomato
-
-cell_9769 cell_9769 AAACCTGAGACTGTAA 5 E8.5 TRUE
-cell_9770 cell_9770 AAACCTGAGATGCCTT 5 E8.5 TRUE
-cell_9771 cell_9771 AAACCTGAGCAGCCTC 5 E8.5 TRUE
-cell_9772 cell_9772 AAACCTGCATACTCTT 5 E8.5 TRUE
-cell_9773 cell_9773 AAACGGGTCAACACCA 5 E8.5 TRUE
-... ... ... ... ... ...
-cell_12175 cell_12175 TTTGGTTAGTCCGTAT 5 E8.5 TRUE
-cell_12176 cell_12176 TTTGGTTAGTGTTGAA 5 E8.5 TRUE
-cell_12177 cell_12177 TTTGGTTGTTAAAGAC 5 E8.5 TRUE
-cell_12178 cell_12178 TTTGGTTTCAGTCAGT 5 E8.5 TRUE
-cell_12179 cell_12179 TTTGGTTTCGCCATAA 5 E8.5 TRUE
- pool stage.mapped celltype.mapped closest.cell
-
-cell_9769 3 E8.25 Mesenchyme cell_24159
-cell_9770 3 E8.5 Endothelium cell_96660
-cell_9771 3 E8.5 Allantois cell_134982
-cell_9772 3 E8.5 Erythroid3 cell_133892
-cell_9773 3 E8.25 Erythroid1 cell_76296
-... ... ... ... ...
-cell_12175 3 E8.5 Erythroid3 cell_138060
-cell_12176 3 E8.5 Forebrain/Midbrain/H.. cell_72709
-cell_12177 3 E8.25 Surface ectoderm cell_100275
-cell_12178 3 E8.25 Erythroid2 cell_70906
-cell_12179 3 E8.5 Spinal cord cell_102334
- doub.density sizeFactor my_sum
-
-cell_9769 0.02985045 1.41243 27577
-cell_9770 0.00172753 1.22757 29309
-cell_9771 0.01338013 1.15439 28795
-cell_9772 0.00218402 1.28676 34794
-cell_9773 0.00211723 1.78719 38300
-... ... ... ...
-cell_12175 0.00129403 1.219506 26680
-cell_12176 0.01833074 1.095753 19013
-cell_12177 0.03104037 0.910728 24627
-cell_12178 0.00169483 2.061701 46162
-cell_12179 0.03767894 1.798687 38398
-```
+:::
+
+::::
### The `reducedDims`
@@ -344,7 +285,7 @@ library(scater)
plotReducedDim(sce, "pca.corrected.E8.5", colour_by = "celltype.mapped")
```
- +
+ :::::::::::::::::::::::::::::::::: challenge
diff --git a/md5sum.txt b/md5sum.txt
index cfb3af3..dc3af05 100644
--- a/md5sum.txt
+++ b/md5sum.txt
@@ -4,9 +4,9 @@
"config.yaml" "b0d664d3d6abdd0e98b16282e1c03107" "site/built/config.yaml" "2024-07-02"
"index.md" "495939ddd3f110be3bbcd49b60f4a7ce" "site/built/index.md" "2024-07-02"
"links.md" "8184cf4149eafbf03ce8da8ff0778c14" "site/built/links.md" "2024-07-02"
-"episodes/intro-sce.Rmd" "5d23a77886d651cb1aa1b97a93d2e085" "site/built/intro-sce.md" "2024-08-06"
-"episodes/eda_qc.Rmd" "fac7397fb4caf04a9dc411b5e316ed60" "site/built/eda_qc.md" "2024-07-02"
-"episodes/cell_type_annotation.Rmd" "0aee6fee81ace65c93a9e39b0916a2cb" "site/built/cell_type_annotation.md" "2024-07-02"
+"episodes/intro-sce.Rmd" "43e8d9965c505108e386dffb0f011697" "site/built/intro-sce.md" "2024-08-12"
+"episodes/eda_qc.Rmd" "07eec916cc6aff0c5dcd6ec463b66e75" "site/built/eda_qc.md" "2024-08-12"
+"episodes/cell_type_annotation.Rmd" "fa2333e4c8aa876394327610eb650e8b" "site/built/cell_type_annotation.md" "2024-08-12"
"episodes/multi-sample.Rmd" "75798a2bb3500220ff7b3c1d88cadf41" "site/built/multi-sample.md" "2024-07-02"
"episodes/large_data.Rmd" "6b413ccf15bea3e70c6dd75432d3914d" "site/built/large_data.md" "2024-07-02"
"episodes/hca.Rmd" "09e4390a2d357fdb950d663f3d3fcd7a" "site/built/hca.md" "2024-07-02"
:::::::::::::::::::::::::::::::::: challenge
diff --git a/md5sum.txt b/md5sum.txt
index cfb3af3..dc3af05 100644
--- a/md5sum.txt
+++ b/md5sum.txt
@@ -4,9 +4,9 @@
"config.yaml" "b0d664d3d6abdd0e98b16282e1c03107" "site/built/config.yaml" "2024-07-02"
"index.md" "495939ddd3f110be3bbcd49b60f4a7ce" "site/built/index.md" "2024-07-02"
"links.md" "8184cf4149eafbf03ce8da8ff0778c14" "site/built/links.md" "2024-07-02"
-"episodes/intro-sce.Rmd" "5d23a77886d651cb1aa1b97a93d2e085" "site/built/intro-sce.md" "2024-08-06"
-"episodes/eda_qc.Rmd" "fac7397fb4caf04a9dc411b5e316ed60" "site/built/eda_qc.md" "2024-07-02"
-"episodes/cell_type_annotation.Rmd" "0aee6fee81ace65c93a9e39b0916a2cb" "site/built/cell_type_annotation.md" "2024-07-02"
+"episodes/intro-sce.Rmd" "43e8d9965c505108e386dffb0f011697" "site/built/intro-sce.md" "2024-08-12"
+"episodes/eda_qc.Rmd" "07eec916cc6aff0c5dcd6ec463b66e75" "site/built/eda_qc.md" "2024-08-12"
+"episodes/cell_type_annotation.Rmd" "fa2333e4c8aa876394327610eb650e8b" "site/built/cell_type_annotation.md" "2024-08-12"
"episodes/multi-sample.Rmd" "75798a2bb3500220ff7b3c1d88cadf41" "site/built/multi-sample.md" "2024-07-02"
"episodes/large_data.Rmd" "6b413ccf15bea3e70c6dd75432d3914d" "site/built/large_data.md" "2024-07-02"
"episodes/hca.Rmd" "09e4390a2d357fdb950d663f3d3fcd7a" "site/built/hca.md" "2024-07-02"