Issue with a large Pseudomonas dataset #299
Comments
|
Regarding the QC options, could you try with: It would be useful to know from the output (which I think you should still get) the reasons isolates are being removed Regarding query assignment: this does not update the distance plot but you should still get clusters assignments out. Are you having problems/errors in this output? A flag that can be helpful in these cases is |
|
We tried the extra flags you provided when running the QC, this time it ran to completion, We were wondering, since you used a similar Pseudomonas dataset in the original PopPUNK paper and in bacpop, if you could share:
|

|
@samhorsfield96 I think you created the pseudomonas DB? Do you remember/know if there was anything special you did? We wouldn't have changed the sequence files (e.g. removing plasmids), but we would have removed isolates. It still looks to me like you have a few isolates that need to be removed to get this to work. The other thing that would be useful is to find those which are giving zero core distance. You can do that with a command like: Which should give you the isolates with the most 'bad' distances from most to least |
|
Hi both, The database on the bacpop website was generated using: From memory I had to increase the sketch size and k-mer ranges to increase resolution at smaller core distances. I also reduced the genome size standard deviation boundary to remove likely contaminated genomes. Let me know if you have any further questions. |
About the dataset
We have two datasets, one with 8046 Pseudomonas genomes, and
another, a subset of the first, with 6347 Pseudomonas aeruginosa genomes.
For the issues described here we will focus on the latter, but the issues are common to both.
About the issue
As part of our workflow, we create subsets based on the core and accessory distances, as infered by PopPUNK.
In this dataset we found that many of the distances for the genomes are 0 for both the accessory and the core genomes.
Here are the command we used to extract the distance table:
poppunk --create-dbpoppunk_extract_distances.pydbscanmodel:Here is the resulting figure:
You can see the number of retained genomes is very small compared to the input size (6347), this is something we've only encountered in this dataset.
Other datasets of similar size have yielded more sensible subsets, e.g. an 11373 genome dataset resulted in these subsets (with a few more thresholds displayed to show how it scales):
Some things we've tried
poppunk_assign. This seemingly didn't change anything on the original PopPUNK db.Before adding to the database:
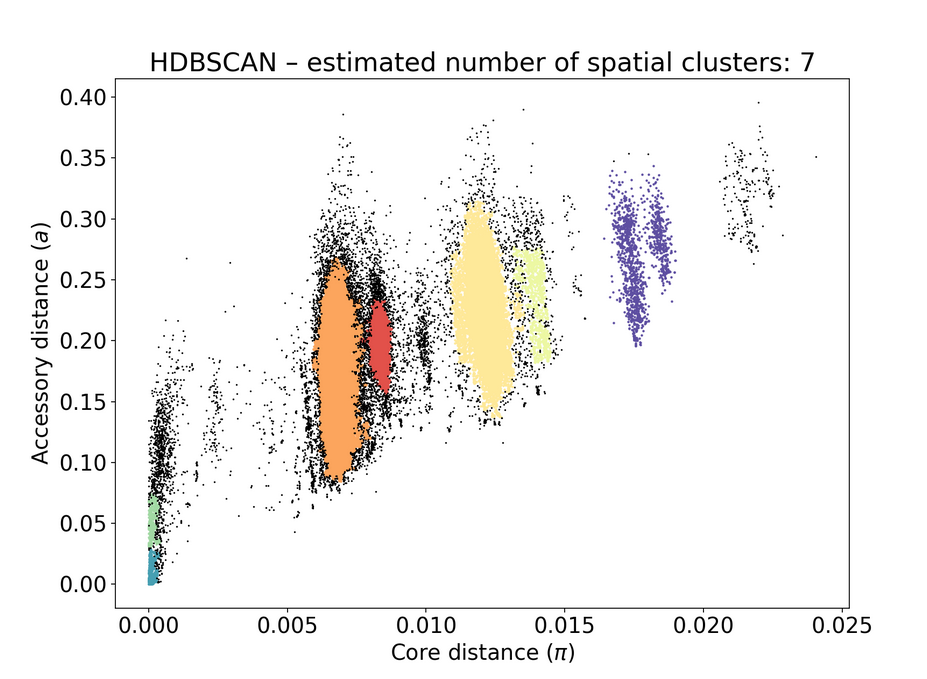
After:
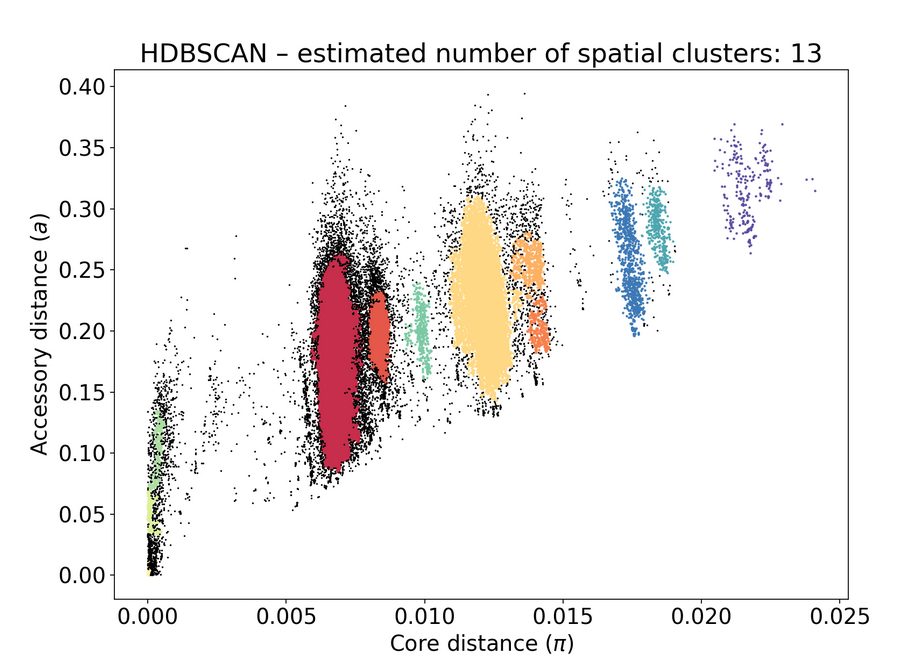
The text was updated successfully, but these errors were encountered: