diff --git a/PIPELINE.md b/PIPELINE.md
new file mode 100644
index 0000000..971b911
--- /dev/null
+++ b/PIPELINE.md
@@ -0,0 +1,45 @@
+# Pipeline Information
+This pipeline is the backbone of sPARcRNA_Viz and provides the coordinates required to create the scRNA-seq visualizations.
+
+## Input
+It takes the barcodes, features, and matrix files as inputs. The files need to either be in .csv/.tsv and .mtx format or in an R data format.
+
+## Output
+A json file with all the coordinates of the points in a tSNE that is used by the frontend to visualize it in an interactive way.
+
+## Workflow
+### 1. Setup
+Load libraries, set options, validate and prepare the directories, find and read raw data files, configure based on inputs
+### 2. Create Seurat object
+Seurat is an R package designed for QC, analysis, and exploration of single-cell RNA-seq data.
+- Seurat was chosen because the gene expression data analyzed through this pipeline is single-cell RNA-seq, and it provides ways to normalize, scale, and visualize this data.
+### 3. Normalize and preprocess the data
+Normalize (so that data reflects true biological differences), find variable features, scale (to standardize the data), perform PCA (Principal Component Analysis to reduce dimensionality), cluster cells with similar profiles together
+### 4. t-SNE
+t-SNE allows us to visualize statistically significant genes based on these clusters. From these, researchers can determine potential gene ontologies arising from their sample(s).
+### 5. Differential Gene Expression Analysis
+Differential gene expression analysis takes the normalized gene read counts and allows researchers to determine quantitative changes in gene expression.
+### 6. GSEA
+GSEA, or Gene set enrichment analysis, helps determine the gene groups that are highly represented in the data.
+### 7. Combine t-SNE and GSEA results
+All the cluster results after running GSEA are saved, and the top pathways are saved as well.
+### 8. Export and Display Results
+All values from the previous steps and top clusters, pathways, etc are saved in a json file that is later visualized
+
+## Overview of Functions
+- `make_options()`: allows for user input through command line, allows to input data files from local machine
+- `Read_MTX()`: reads the data from barcodes, features, and matrix files after patterns have been made and properly found from the input files given by the user
+- `CreateSeuratObject()`: Seurat object created from data saved and user inputs on the name, cells, and features
+- Cleaning the data and making it standardized so that it can be used for a tSNE and GSEA:
+ - `NormalizeData()`
+ - `ScaleData()`
+- Reducing the dimensionality, clustering, and running the tSNE and saving it:
+ - `RunPCA()`
+ - `FindNeighbors()`
+ - `FindClusters()`
+ - `RunTSNE()`
+ - `DimPlot()`
+ - `ggsave()`
+- `FindAllMarkers()`: performs the differential expression analysis
+- `GetAssayData()`: saves the normalized gene expression data, which makes sure that the data is not due to technical biases
+- tSNE coordinates, top 10 markers, top pathways, cluster results, cluster centroids, cluster average expression data, and more are saved and exported as a json file
diff --git a/README.md b/README.md
index 0871510..029ee0c 100644
--- a/README.md
+++ b/README.md
@@ -1,45 +1,211 @@
-# osparc_dex_service
-Easily generate differential expression results from OSparc data
+
+  +
+  +
+
+  +
+
+
-## Requirements
+# Table of Contents
+- [About](#about)
+- [Research Purpose](#research-purpose)
+ - [Introduction](#introduction)
+ - [Background](#background)
+ - [Current SPARC Portal Tools](#current-sparc-portal-tools)
+ - [The Problem](#the-problem)
+ - [Our Solution: sPARcRNA_Viz](#our-solution-sparcrna_viz)
+- [Using sPARcRNA_Viz](#using-sparcrna_viz)
+ - [sPARcRNA_Viz Requirements](#sparcrna_viz-requirements)
+ - [sPARcRNA_Viz Pipeline Workflow](#sparcrna_viz-pipeline-workflow)
+ - [Configuring sPARcRNA_Viz](#configuring-sparcrna_viz)
+ - [Tutorial](#tutorial)
+- [FAIR-Centered Design](#fair-centered-design)
+ - [Importance of FAIR Data Principles](#importance-of-fair-data-principles)
+- [Additional Information](#additional-information)
+ - [Issue Reporting](#issue-reporting)
+ - [How to Contribute](#how-to-contribute)
+ - [Cite Us](#cite-us)
+ - [License](#license)
+ - [Team](#team)
+ - [Materials Cited](#materials-cited)
+ - [Acknowledgements](#acknowledgements)
+
+# About
+Easily generate differential expression results from [SPARC](https://sparc.science) scRNA-seq data in a FAIR manner.
+# Research Purpose
+## Introduction
+sPARcRNA_Viz is an **all-in-one gene expression visualization utility** integratable with [o²S²PARC](https://osparc.io/). Using sPARcRNA_Viz, researchers can create an interactive t-SNE from single-cell RNA-sequencing data, as well as perform in silico GSEA analysis to determine the most highly expressed genes. From these statistically significant genes, researchers can determine potential gene ontologies arising from their sample(s). In addition, the seamless integration of sPARcRNA_Viz with the o²S²PARC computing platform enables data accessibility concordant with **FAIR Data Principles**.
+### Notable Features of sPARcRNA_Viz
+sPARcRNA_Viz provides the user with the ability to fine-tune multiple **gene expression parameters**:
+- Minimum number of cells expressing a gene
+- Minimum number of features (genes) per cell
+- Maximum number of features (genes) per cell
+- Clustering resolution
+- Species specification
+- Minimum percentage for FindAllMarkers
+- Log fold-change threshold for FindAllMarkers
+- Minimum gene set size for GSEA
+- MSigDB category for GSEA
+### Technology Stack
+- o²S²PARC
+- R
+- GNU Make
+- Python3
+- Docker
+- Astro
+- HTML
+- JavaScript
+- Tailwind CSS
+## Background
+In recent years, **single-cell RNA-sequencing** (scRNA-seq) has emerged as a preeminent method for the analysis of gene expression in biological tissue, providing researchers access to genetic data previously inaccessible. This is largely due to advancements in wet lab and dry leb techniques, as well computing power, where these improvements enable the collection of large datasets often spanning hundreds of millions of entries. With this newfound wealth of data, a need has arisen for high-efficiency bioinformatics pipelines and tools that allow for the analysis of scRNA-seq data. One computational method currently in use is **differential gene expression (DGE) analysis**, which identifies statistically significant genes (i.e., results that are minimally confounded by experimental errors) and determines the expression level of a gene relative to the entire dataset.2 Using these statistically significant results, it is possible to correlate the most highly expressed genes to their tangible, biological effects through the use of **gene ontology** databases such as the [Gene Ontology Knowledgebase (GO)](https://www.geneontology.org/).
+
+The SPARC Portal currently hosts a rich collection of scRNA-seq data across several different tissues and species. Therefore, the SPARC platform could be further enhanced by the inclusion of data visualization and the aforementioned DGE tools. This is achieved in sPARcRNA_Viz through the use of **t-SNE plotting** and **GSEA**.
+### About t-SNE Plots
+t-distributed Stochastic Neighbor Embedding (t-SNE) is a plotting and visualization technique that focuses on pairwise similarities among datasets. Like PCA, it is a dimensionality reduction technique. For its utility in comparing large, complex datasets, t-SNE is commonly employed by RNA-seq researchers.
+### About GSEA
+Gene Set Enrichment Analysis (GSEA) is a popular technique for determining statistically significant genes, as well as those that are upregulated and downregulated.5 This is achieved through a ranking system whereby genes are organized by statistically significance.
+
+## Current SPARC Portal Tools
+As of 8/12/24, the [Transcriptomic_oSPARC](https://github.com/SPARC-FAIR-Codeathon/Transcriptomic_oSPARC) utility1 would appear to be the most prominent SPARC tool relating to the analysis gene expression. This tool is very effective in displaying industry-standard static graphical outputs, which can prove quite useful to researchers. However, a limitation may perhaps exist in the current customization level; it may be necessary to edit the code itself to change particular parameters. There was also a niche to explore in adding interactivity to the graphs, further enahncing the user experience.
+## The Problem
+The gene expression data in SPARC is somewhat limited and is in a raw data format, rendering it less interoperable. Our goal was to make it more interoperable and easy to use. Therefore, our team sought to create a RNA-seq visualization utility that supports the specification of **specific parameters**, as well as **interactivity**. There was also room for experimentation in predicting gene ontology with **GSEA**.
+## Our Solution: sPARcRNA_Viz
+To address this challenge, we present **sPARcRNA_Viz**, an scRNA-seq visualization tool for potential entry alongside Transcriptomic_oSPARC. In incorporating flexible parameters, interactivity, and an additional DEA metric, sPARcRNA_Viz will complement Transcriptomic_oSPARC as part of a growing SPARC gene expression toolkit.
+
+# Using sPARcRNA_Viz
+## sPARcRNA_Viz Requirements
- GNU Make
- Python3
- [``Docker``](https://docs.docker.com/get-docker/) (if you wish to build and test the service locally)
+### Required Input Format
+sPARcRNA_Viz currently supports the following file format: **.csv/.tsv** (barcode and feature files), **.mtx** (matrix file) single-cell matrices along with R data. These formats and 3 files are required to run the analysis successfully.
+## sPARcRNA_Viz Pipeline Workflow
+Can refer to [PIPELINE.md](PIPELINE.md).
+### 1. Setup
+Load libraries, set options, validate and prepare the directories; find and read raw data files; configure based on inputs.
+### 2. Create Seurat object
+[Seurat](https://cran.r-project.org/web/packages/Seurat/index.html) is an R package specially designed for the quality control (QC) , analysis, and exploration of single-cell RNA-seq data. Thus, it proved to be a suitable choice for the purposes of sPARcRNA_Viz.
+### 3. Normalize and preprocess the data
+Normalize (so that data reflects true biological differences); find variable features; scale (to standardize the data); perform PCA (Principal Component Analysis to reduce dimensionality); and cluster cells with similar profiles together.
+### 4. t-SNE
+t-SNE allows us to visualize statistically significant genes based on these clusters. From these, researchers can determine potential gene ontologies arising from their sample(s).
+### 5. Differential Gene Expression Analysis
+Differential gene expression analysis takes the normalized gene read counts and allows researchers to determine quantitative changes in gene expression.
+### 6. GSEA
+GSEA aids in determining gene groups highly represented in the data.
+### 7. Combine t-SNE and GSEA results
+All the cluster results after running GSEA are saved, and the top pathways are saved as well.
+### 8. Export and Display Results
+All values from the previous steps and top clusters, pathways, etc are saved in a Seurat object that is later visualized. The user can optionally convert this data into .csv file format.
+
+## Configuring sPARcRNA_Viz
+sPARcRNA_Viz offers a variety of command options:
+| Option | Description | Default |
+| --- | --- | --- |
+| `-i`, `--input` | Input directory path | `'validation/input/data/'` |
+| `-o`, `--output` | Output directory path | `'tmp_output/'` |
+| `-n`, `--name` | The name of the dataset being analyzed | `"sPARcRNA"` |
+| `-c`, `--min_cells` | Minimum number of cells expressing a gene | `3` |
+| `-f`, `--min_features` | Minimum number of features (genes) per cell | `200` |
+| `--max_features` | Maximum number of features (genes) per cell | `2500` |
+| `--resolution` | Resolution parameter for clustering | `0.8` |
+| `--species` | Species for GSEA | `"Mus musculus"` |
+| `--min_pct` | Minimum percentage for FindAllMarkers | `0.25` |
+| `--logfc_threshold` | Log fold-change threshold for FindAllMarkers | `0.25` |
+| `--gsea_min_size` | Minimum gene set size for GSEA | `15` |
+| `--gsea_max_size` | Maximum gene set size for GSEA | `500` |
+| `--category` | MSigDB category for GSEA | `"H"` |
+## Tutorial
+The [scRNA-seq data](https://sparc.science/datasets/220?type=dataset&datasetDetailsTab=files&path=files/derivative) used in the tutorial is from the SPARC Portal.
+### 1. Log in to [o²S²PARC](https://osparc.io/)
+ +
+### 2. Open a new Study
+
+
+### 2. Open a new Study
+ +
+### 3. Add 3 File Picker Nodes and upload the required data
+
+
+### 3. Add 3 File Picker Nodes and upload the required data
+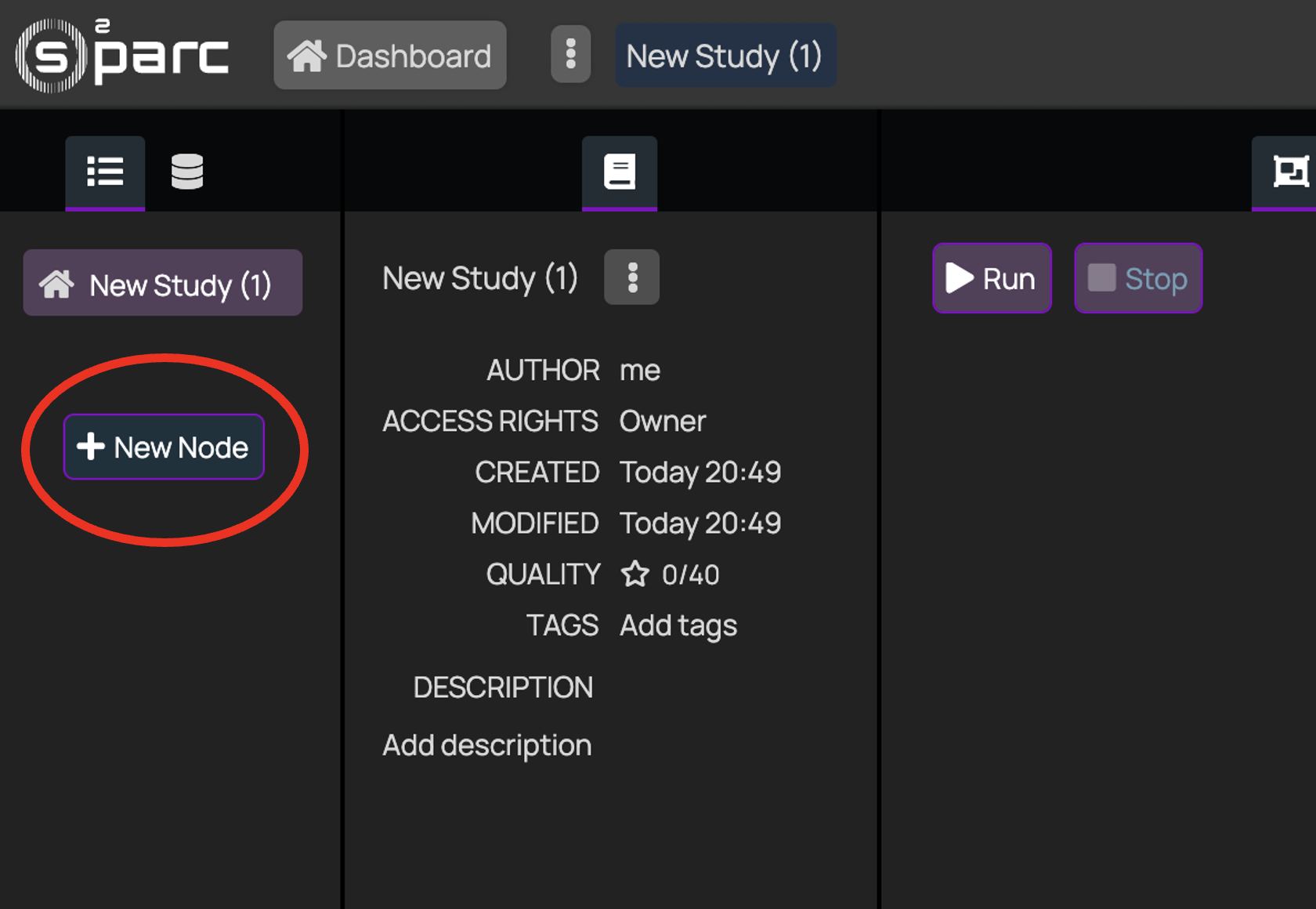 +
+
+ +
+
+ +
+
+(Alternatively, drag and drop the needed files into the workspace.)
+
+### 4. Add sPARcRNA_Viz Node
+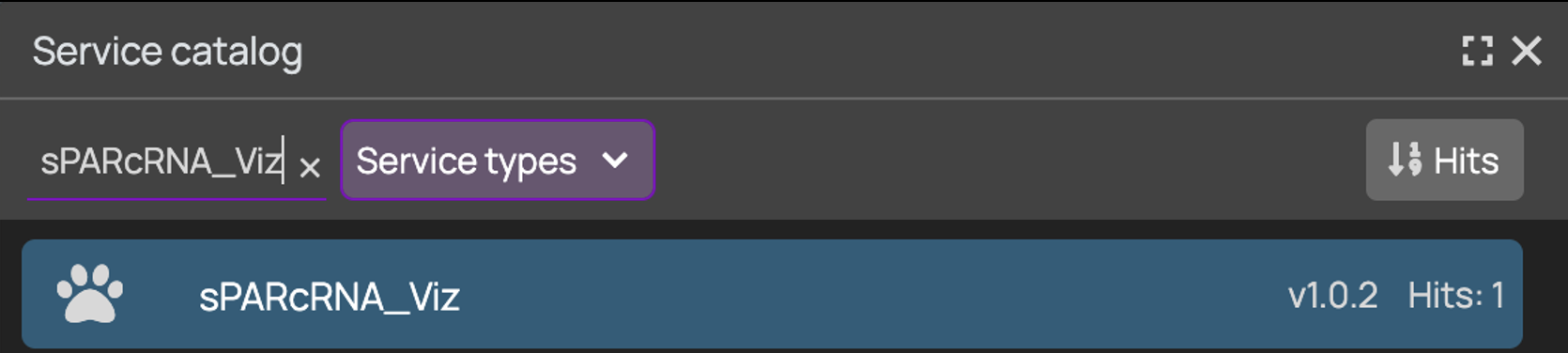 +
+### 5. Connect the Nodes
+
+
+### 5. Connect the Nodes
+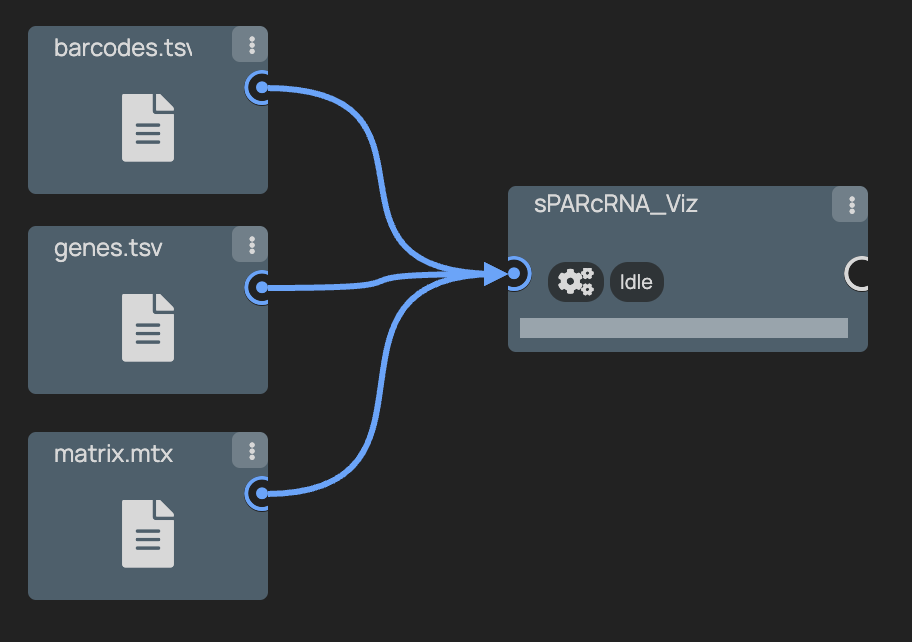 +
+### 6. Optionally run outputs through JupyterLab R for further analysis
+
+
+### 6. Optionally run outputs through JupyterLab R for further analysis
+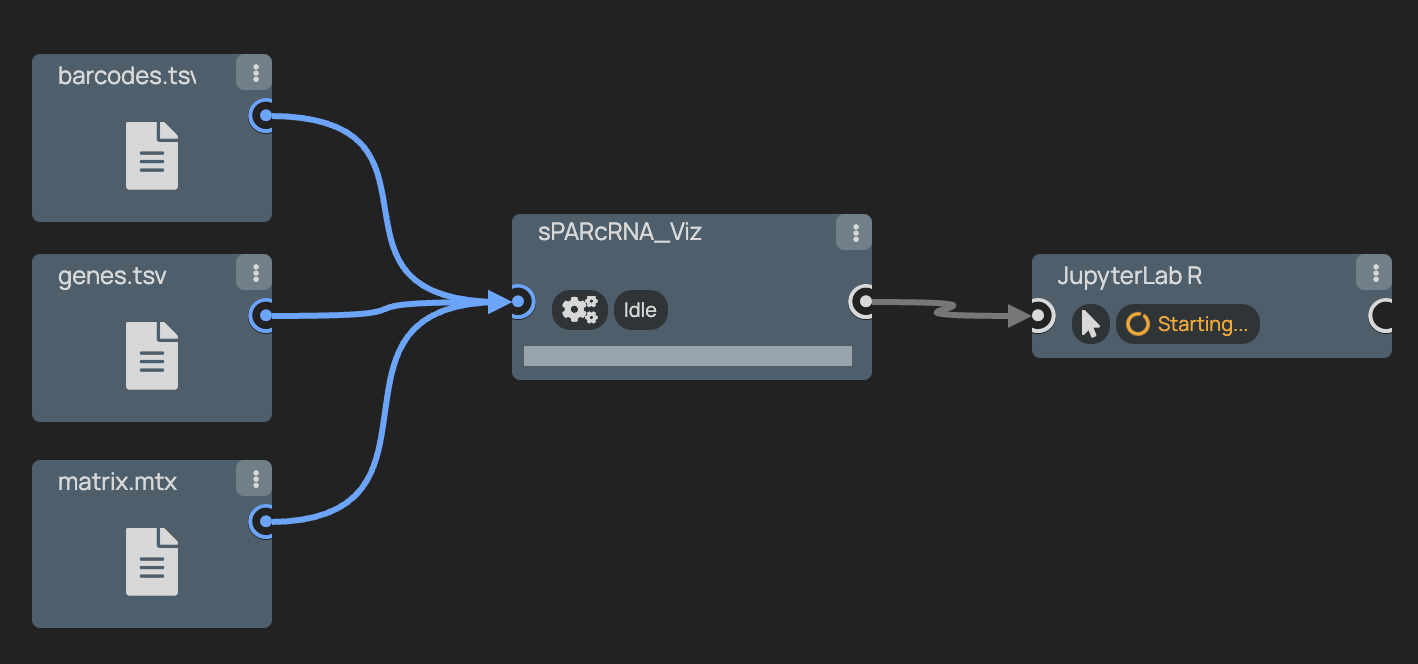 +
+## Future Vision
+sPARcRNA_Viz would be expanded to include other interactive visualizations and API calls to other gene databases. This would provide more ways to analyze genes and integrate with other websites.
+
+# FAIR-Centered Design
+Perhaps the **most important** aspect of sPARcRNA_Viz is its emphasis on the FAIR Data Principles. Summarized below are highlight features of sPARcRNA_Viz supporting the FAIR initiative.
+## Importance of FAIR Data Principles
+
+
+## Future Vision
+sPARcRNA_Viz would be expanded to include other interactive visualizations and API calls to other gene databases. This would provide more ways to analyze genes and integrate with other websites.
+
+# FAIR-Centered Design
+Perhaps the **most important** aspect of sPARcRNA_Viz is its emphasis on the FAIR Data Principles. Summarized below are highlight features of sPARcRNA_Viz supporting the FAIR initiative.
+## Importance of FAIR Data Principles
+
+  +
+
+
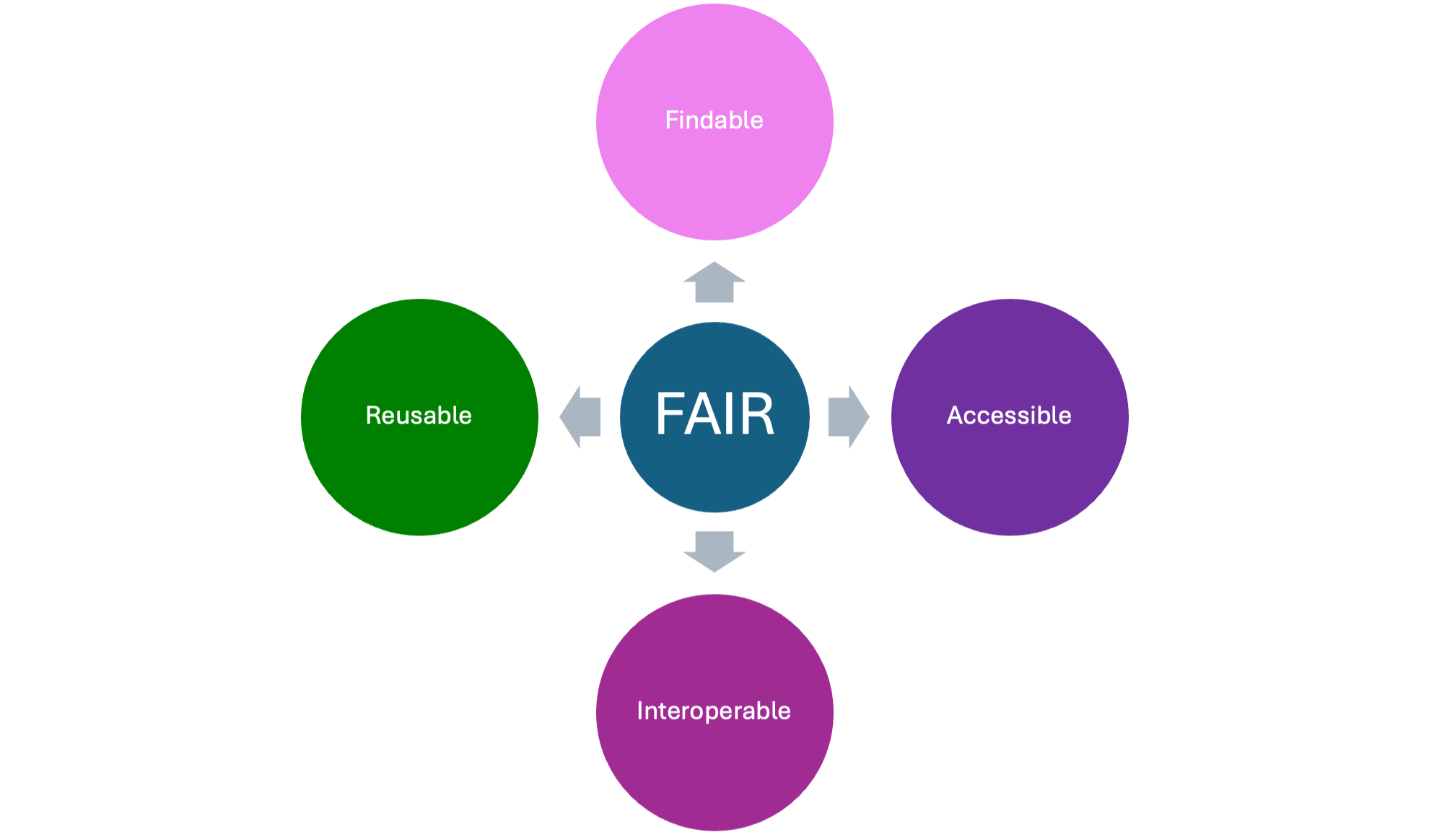
+FAIR data is that which is **F**indable, **A**ccessible, **I**nteroperable, and **R**eusable.3 These tenets are highly important within the scientific context as a lack of clarity surrounding data may introduce confounding variables. Thus, the FAIR system was derived so as to create a method to organize data, thus serving both present and future researchers.
+
+Particularly in the case of scRNA-seq data, which is expensive from both a wet and dry lab standpoint, it is very useful to adhere to FAIR standards. For instance, one particularly common phenomemon with respect to scRNA-seq is **dropout**4, where portions of RNA are not captured by experimental techniques. scRNA-seq data can also be signficantly varied with regard to format; often, differently-labeled matrices may contain raw counts data, or data that has been normalized by a method such as CPM, TPM, or RPKM/FPKM. The FAIR article cited on the SPARC website expands upon this idea further: the licensing of data can also pose a challenge for the analysis of gene regulation and expression. Therefore, the intentional **categorization and stewardship** of data can present a major benefit to transcriptomics researchers, propelling scientific progress.
+
+### Summary of FAIR Principles Application
+| FAIR Principle | Other Tools | sPARcRNA_Viz |
+| --- | --- | --- |
+| **F**indable | May not be connected to an existing database such as the SPARC Portal, which could hinder the findability of data | sPARcRNA_Viz is **connected to o²S²PARC**, so it can use the well-organized datasets provided on the SPARC portal, and it is archived on Zenodo with the appropriate metadata |
+| **A**ccessible | May have a user interface that requires a programming background | sPARcRNA_Viz's **friendly user interface and visuals** allow researchers to quickly engage with data and is open, free and universally implementable |
+| **I**nteroperable | May not allow for connections between datasets | Through its use of GSEA, sPARcRNA_Viz allows for the **meaningful connection of datasets**: scRNA-seq data can be used in association with gene ontology. In addition, visualizations generated for each dataset can be compared with each other |
+| **R**eusable | May only support the formatting of one dataset | sPARcRNA_Viz be used with multiple datasets due to the ability to **specify parameters**. Likewise, sPARcRNA_Viz offers a security benefit through its use of **input validation** |
-## Workflow
-### Create the Service
-1. The [Dockerfile](osparc_dex_service/docker/Dockerfile) shall be modified to install the command-line tool you'd like to execute and additional dependencies
-
-All the rest is optional:
-1. The [.osparc](.osparc) is the configuration folder and source of truth for metadata: describes service info and expected inputs/outputs of the service. If you need to change the inputs/outputs of the service, description, thumbnail, etc... check the [`metadata.yml`](./.osparc/metadata.yml) file
-2. If you need to change the start-up behavior of the service, modify the [`service.cli/execute.sh`](./service.cli/execute.sh) file
-
-
-Testing:
-1. The service docker image may be built with ``make build`` (see "Useful Commands" below)
-2. The service docker image may be run locally with ``make run-local``. You'll need to edit the [input.json](./validation/input/inputs.json) to execute your command.
-
-### Publish the Service on o²S²PARC
-Once you're happy with your code:
-1. Push it to a public repository.
-2. An automated pipeline (GitHub Actions) will build the Docker image for you
-3. Wait for the GitHub pipeline to run successfully
-4. Check that the automated pipeline executes successfully
-5. Once the pipeline has run successfully, get in touch with [o²S²PARC Support](mailto:support@osparc.io), we will take care of the final steps!
-
-### Change the Service (after it has been published on o²S²PARC )
-If you wish to change your Service (e.g. add additional libraries), after it has been published on o²S²PARC, you have to **create a new version**:
-1. Go back to your repository
-2. Apply the desired changes and commit them
-3. Increase ("bump") the Service version: in your console execute: ``make version-patch``, or ``make version-minor``, or ``make version-major``
-4. Commit and push the changes to your repository
-5. Wait for the GitHub/GitLab pipelines to run successfully
-5. Once the pipeline has run successfully, get in touch with [o²S²PARC Support](mailto:support@osparc.io), we will take care of publishing the new version!
-
-
-### Useful commands
-```console
-$ make help
-$ make build # This will build an o²S²PARC-compatible image (similar to `Docker build` command)
-$ make run-local # This will start a new Docker container on your computer and run the command.
\ No newline at end of file
+# Additional Information
+## Issue Reporting
+Please utilize the **Issues** tab of this repository should you encounter any problems with sPARcRNA_Viz.
+## How to Contribute
+Please Fork this repository and submit a **Pull Request** to contribute.
+## Cite Us
+Please see our [citation](CITATION.cff).
+## License
+sPARcRNA_Viz is distributed under the [MIT License](LICENSE).
+## Team
+- Mihir Samdarshi (Lead, Sysadmin, Developer)
+- Sanjay Soundarajan (Sysadmin, Developer)
+- Mahitha Simhambhatla (Developer, Writer)
+- Raina Patel (Writer)
+- Ayla Bratton (Writer)
+## Materials Cited
+[1]
+Ben Aribi, H., Ding, M., & Kiran, A. (2023).
+Gene expression data visualization tool on the o2S2PARC platform.
+F1000Research, 11, 1267.
+https://www.pnas.org/doi/abs/10.1073/pnas.0506580102
+[2]
+EMBL-EBI. (n.d.).
+Differential gene expression analysis | Functional genomics II.
+https://www.ebi.ac.uk/training/online/courses/functional-genomics-ii-common-technologies-and-data-analysis-methods/rna-sequencing/performing-a-rna-seq-experiment/data-analysis/differential-gene-expression-analysis/
+[3]
+GO FAIR.(2017).
+FAIR Principles - GO FAIR. GO FAIR.
+https://www.go-fair.org/fair-principles/
+[4]
+Kim, T. H., Zhou, X., & Chen, M. (2020). Demystifying “drop-outs” in single-cell UMI data. Genome Biology, 21(1).
+https://doi.org/10.1186/s13059-020-02096-y
+[5]
+Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., & Mesirov, J. P. (2005).
+Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles.
+Proceedings of the National Academy of Sciences, 102(43), 15545–15550.
+https://doi.org/10.1073/pnas.0506580102
+[6]
+Wilkinson, M. D., Dumontier, M., Aalbersberg, Ij. J., Appleton, G., Axton, M., Baak, A., Blomberg, N., Boiten, J.-W., da Silva Santos, L. B., Bourne, P. E., Bouwman, J., Brookes, A. J., Clark, T., Crosas, M., Dillo, I., Dumon, O., Edmunds, S., Evelo, C. T., Finkers, R., & Gonzalez-Beltran, A. (2016).
+The FAIR Guiding Principles for Scientific Data Management and Stewardship. Scientific Data, 3(1).
+https://www.nature.com/articles/sdata201618
+
+Logo and figures were created using Microsoft Word; images were formatted using Canva.
+## Acknowledgements
+We would like to thank the SPARC Codeathon 2024 team for all their guidance and support.